


HIV/AIDS: Relato sobre a epidemia, terapias anti-retrovirais disponíveis e fatores relacionados à aq
RESUMO
Existem no mundo aproximadamente 33 milhões de pessoas vivendo com HIV/AIDS, destas, cerca de 474 mil vivem no Brasil. Apesar do crescimento da epidemia no mundo, o desenvolvimento dos anti-retrovirais (ARV) mudou a vida dos indivíduos vivendo com HIV/AIDS. A morbidade e mortalidade declinaram entre 60 e 80%, principalmente pela disponibilidade de diferentes classes de fármacos ARV e o uso em combinação de três ou mais deles. O conhecimento da dinâmica viral e o surgimento de métodos laboratoriais capazes de mensurar a quantidade de vírus circulante no plasma tornaram possível a monitorização confiável e objetiva da evolução clínica e do tratamento da infecção pelo HIV. No entanto, apesar dos vários fármacos disponíveis, até mesmo pacientes virgens de tratamento podem apresentar resistência aos ARV. Neste trabalho os autores fizeram uma revisão da literatura no sentido de avaliar as principais mutações relacionadas à resistência viral as diferentes classes de ARV. Este estudo reforça a necessidade da adesão ao tratamento como forma de evitar a replicação viral e conseqüente favorecimento de mutações de resistência.
Palavras-chave: Anti-retrovirais, HIV, Resistência, HAART, Genotipagem
LISTA DE ABREVIATURAS E SIGLAS
3TC – Lamivudina
ABC – Abacavir
AIDS - Síndrome de imunodeficiência adquirida
ARV – Anti-retroviral
ATV – Atazanavir
ATP – Adenosina trifosfato
ASC – Área sob a curva
AZT – Zidovudina
CYP450 – Citocromo P450
d4T – Estavudina
ddC – Zalcitabina
ddI – Didanosina
DNA – Ácido Desoxirribonucléico
EFZ – Efavirenz
FDA - Food and Drug Administration
FPV – Fosamprenavir
FTC – Emtricitabina
HAART – Terapia anti-retroviral altamente eficaz
HBV – Vírus da hepatite B
HIV – Vírus da imunodeficiência Humana
IDV – Indinavir
IMC – Índice de massa corporal
IP - Inibidores da protease
IsF – Inibidores de Fusão
ITRN - Inibidores da trancriptase reversa análogos de nucleosídeos
ITRNN - Inibidores da transcriptase reversa não análogos de nucleosídeos
LCR – LÍquido cefalorraquidiano
LPV/r – Lopinavir
LT – Linfócitos T
LTR - Long terminal repeats
MHC - Complexo principal de histocompatibilidade
NVP – Nevirapina
OMS – Organização mundial de saúde
RNA – Ácido Ribonucléico
RTV – Ritonavir
SNC – Sistema nervoso Central
SQV – Saquinavir
T-20 – Enfuvirtida
TARV – Terapia Anti-retroviral
TDF - Tenofovir disoproxil fumarato
TR – Trascriptase reversa
1 INTRODUÇÃO
A síndrome de imunodeficiência adquirida (AIDS) foi descrita pela primeira vez no inicio da década de oitenta em Nova York nos EUA (RAMOS NETO, 2004). Grupos de pacientes jovens, homossexuais masculinos, exibiam um complexo de sintomas, incluindo pneumonia severa causada por Pneumocystis jiroveci (STRINGER et al., 2002; STROHL et al., 2004) (normalmente um organismo eucariótico inofensivo), sarcoma de Kaposi (ordinariamente uma forma extremamente rara de câncer), perda de peso súbita, linfadenopatia e supressão geral da função imune. Esse conjunto de sinais e sintomas associados à doença veio a ser conhecido como a síndrome de imunodeficiência adquirida (STROHL et al., 2004).
Atualmente, acredita-se que o HIV tenha surgido pela mutação de um vírus que era endêmico em algumas áreas da África Central por muitos anos. Os pesquisadores especulam que um vírus relativamente benigno infectando macacos penetrou na população humana quando os animais eram mortos, sua pele retirada e sua carne utilizada para alimentos (TORTORA et al., 2005).
O HIV é um retrovírus do gênero Lentivirus, que possui duas fitas idênticas de RNA, a enzima transcriptase reversa (TR) e um envelope fosfolipídico (TORTORA et al., 2005). Os genes podem ser divididos em dois grupos: os que codificam as proteínas estruturais (gag, pol e env) e os que codificam proteínas não-estruturais regulatórios (tat, rev), necessários para replicação viral in vitro, e genes acessórios (nef, vif, vpu e vpr), que não são essenciais (FOCACCIA & VERONESI, 2007). A integrase, RNAse, protease e transcriptase reversa, tão importante para que ocorra o ciclo viral na célula hospedeira, tratam-se de um complexo protéico pertencente ao gene pol do HIV (BISMARA, 2006). O envelope apresenta em sua superfície uma membrana lipídica oriunda da membrana externa da célula do hospedeiro e duas glicoproteínas (gp41 e gp120) (FOCACCIA & VERONESI, 2007). O envelope envolve o nucleocapsídeo viral de formato cônico. Apresenta um genoma de aproximadamente 10kb, que codifica diversas proteínas, como as estruturais do core ou cerne viral e as com atividade enzimática (BISMARA, 2006).
Dois tipos virais distintos do HIV foram identificados. HIV-1 é o tipo associado com a doença nos Estados Unidos, Europa, África Central, e outras partes do mundo (KLIMAS et al., 2008), na classificação através de analises filogenéticas estão distribuídos nos Grupos M (major) (CAVALCANTI, 2005; MORALES et al., 2005), O (outlier), e o N (non-N, non-O). O grupo M é o mais prevalente e subdividido em 11 subtipos (A1, A2, B, C, D, F1, F2, G, H, J, K) (CAVALCANTI, 2005) e 43 recombinantes (LOS ALAMOS, 2008). HIV-2 foi encontrado principalmente em indivíduos infectados na parte ocidental da África e é muito semelhante ao HIV-1, na medida em que tem o mesmo tropismo por células do sistema imunológico e provoca a doença que resulta da deficiência imunológica (KLIMAS et al., 2008). O HIV-2 pode ser dividido em 7 subtipos de A – G (LORETE, 2005).
Assim como outros vírus RNA, o HIV tem alta variabilidade genética. Dentre os mecanismos responsáveis pela geração de variabilidade esta a transcriptase reversa, que incorpora erroneamente em torno de 10-4 bases em cada ciclo replicativo. Como o HIV tem 104 pares de bases em seu genoma, pode-se dizer que ocorre uma substituição nucleotídea por genoma a cada ciclo replicativo, fazendo com que a população de retrovírus contenha pouco ou nenhum genoma idêntico. Por este motivo, o HIV é considerado uma “quasispécie” (FOCACCIA & VERONESI, 2007).
A interação do HIV com o sistema imunológico do hospedeiro é complexa. O vírus utiliza proteínas de superfície das células do sistema imune para penetração. A infecção inicia-se com a penetração do vírus na célula por meio da ligação da proteína gp120 com a molécula CD4 (FOCACCIA & VERONESI, 2007) que é expressa em Linfócitos T auxiliares (LT CD4+), macrófagos e células dendríticas (BISMARA, 2006). Por esta razão, o quadro clinico da AIDS é caracterizado em função da contagem sanguínea de LT CD4+ no individuo infectado, e da caracterização das condições clínicas relacionadas à infecção com o HIV (PEÇANHA et al., 2002).
Posteriormente foi descoberto que dependendo da seqüência primária da sua gp120, diferentes estirpes de HIV-1 podem usar CCR5 (CD195 - receptores de MIP-1 (CCL3), MIP-1 (CCL4), RANTES (CCL5), CCR3 (receptor de Eotaxina (CCL11), RANTES, MIP-1, MCP-3 (CCL7), MCP-4 (CCL13), ou CXCR4 (CD184 – receptor de SDF-1 (CXCL12) (PEREIRA & PEREIRA, 2001; IUIS/WHO, 2001), ou uma combinação desses receptores de quimiocina para entrar na célula alvo. Curiosamente, alguns indivíduos expostos ao HIV-1 permanecem não-infectados ou nunca mostram soro conversão. Embora uma razão para isto deve-se a ausência de uma molécula CCR5 funcional, algumas pessoas parecem montar uma resposta imune humoral muito eficaz que inclui anticorpos IgA contra a gp41 e IgG reconhecendo um complexo gp120 - CD4 (KAUL & LIPTON, 2006). Com o conhecimento de que o vírus da imunodeficiência humana (HIV) usa o receptor 5 das quimiocinas CC (CCR5) para sua entrada nas células humanas, estão sendo desenvolvidas estratégias que atuem no CCR5 para prevenir e tratar a infecção pelo HIV (LEDERMAN et al., 2006).
Após a identificação do agente causador da AIDS, os avanços mais expressivos têm ocorrido no desenvolvimento de fármacos anti-retrovirais (ARV) efetivos para o tratamento dos indivíduos infectados com HIV (DOMINGOS, 2006).
A terapia anti-retroviral (TARV) iniciou-se em 1986 com o uso da zidovudina (AZT), inibidor da transcriptase reversa do HIV, testada previamente em doenças oncológicas (LOPES, 2007). Na atualidade, dispõe-se de um numero grande e crescente de agentes anti-retrovirais para o tratamento de pacientes infectados principalmente por HIV-1 (KATZUNG, 2006). Todas as etapas no ciclo de replicação do HIV são alvos potenciais para uma droga antiviral (STROHL et al., 2004).
É evidente a eficácia terapêutica, principalmente após a introdução do conceito da HAART (Highly Active Antiretroviral Therapy – Terapia Anti-retroviral Altamente Eficaz), que é a combinação dos inibidores de protease e transcriptase reversa, de forma a ser extremamente efetiva na redução da carga viral plasmática de RNA - HIV-1 para níveis indetectáveis (COLOMBRINI et al., 2006). No entanto, existem importantes barreiras no sucesso a médio e longo prazo do tratamento. As principais dificuldades estão associadas com a toxicidade dos medicamentos, adesão do paciente e resistência viral (FALCI et al., 2006).
A não adesão ao tratamento com a associação dos ARV, tem sido considerada como um dos mais ameaçadores perigos para a efetividade do tratamento, na dimensão individual, e para a disseminação da resistência viral, em nível coletivo (BRITO et al., 2006). A resistência é dividida em primária e secundária: A primária é a que já esta presente mesmo antes do uso da medicação pelo individuo infectado; A secundária é aquela que aparece em conseqüência da pressão seletiva exercida pela TARV (CAVALCANTI, 2005).
Pacientes primariamente infectados com cepas de HIV-1 resistentes ao AZT foram identificados já em 1993, seis anos após a sua introdução como agente ARV (KALMAR, 2007). A partir de 2003, a publicação de testes de resistência medicamentosa tornou-se generalizada no mundo desenvolvido e tem sido aceita como um importante suplemento para a gestão de doentes com viremia detectável no plasma que estão a receber TARV. Além disso, a transmissão de pessoa a pessoa de vírus resistentes aos medicamentos, ocorre em uma variedade de aspectos, incluindo entre adultos e de mãe para o filho, indicando que os testes de resistência aos medicamentos antes de iniciar a terapia pode ser útil até mesmo para pacientes virgens de tratamento. Novas mutações resistentes que conferem resistência a medicamentos mais velhos continuam a ser identificado a cada dia (HIRSCH et al., 2008).
Portanto torna-se imprescindível o desenvolvimento de novas alternativas de tratamentos anti-retrovirais contra HIV, além da realização de testes de resistência, para que se obtenha a diminuição da taxa de mutação e a cura definitiva do vírus HIV.
2 OBJETIVOS
2.1 OBJETIVOS GERAIS
O objetivo do presente trabalho é realizar uma revisão bibliográfica sobre os anti-retrovirais, abordando desde os mecanismos de ação até a aquisição de resistência e os fatores relacionados que levam a falha terapêutica.
2.2 OBJETIVOS ESPECÍFICOS
• Esclarecer os mecanismo de ação dos fármacos Anti-retrovirais;
• Apontar os códons de mutação que levam a uma resistência viral;
• Abordar as causas da falha terapêutica.
3 METODOLOGIA
Através de pesquisa bibliográfica em diferentes bases de dados (Pubmed, Scielo, Ibict, Oaister) e em livros foram selecionados cerca de 60 artigos atualizados sobre o tema. A partir da leitura dos artigos selecionados e capítulos de livros, preparou-se uma monografia de revisão abordando principalmente as opções terapêuticas disponíveis, as reações adversas e interações medicamentosas mais freqüentes, bem como os fatores determinantes para geração de resistência aos anti-retrovirais.
4 FUNDAMENTAÇÃO TEÓRICA
4.1 HIV/AIDS
4.1.1 Epidemiologia
Conforme o relatório anual do Programa Conjunto das Nações Unidas sobre HIV/AIDS existem no mundo aproximadamente 33 milhões de pessoas vivendo com a pandemia (UNAIDS/WHO, 2007).
A África Subsaariana é a área mais afetada, com aproximadamente dois terços do total mundial (22,5 milhões de pessoas com o HIV); desse número três quartos são do sexo feminino. A região também concentra 76% das mortes pela doença. Na América Latina, o relatório afirma que a epidemia permanece estável estimando que 1,6 milhão de pessoas vivam com AIDS. O documento também indica aumento de 150% no número de pessoas infectadas na Europa Oriental e Ásia Central: passou de 630 mil, em 2001, para 1,6 milhão, em 2007. Noventa por cento das pessoas com HIV no Leste Europeu vivem na Ucrânia e na Rússia (UNAIDS/WHO, 2007).
Figura 1: Estimativa dos casos de HIV em Adultos e Crianças no Mundo em 2007.
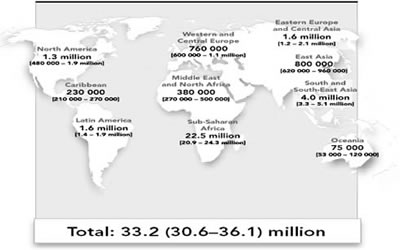
Fonte: (UNAIDS/WHO, 2007)
De 1980 a junho de 2007 foram notificados 474 mil casos de AIDS no Brasil – 289 mil no Sudeste, 89 mil no Sul, 53 mil no Nordeste, 26 mil no Centro Oeste e 16 mil no Norte. Nas regiões Sul, Sudeste e Centro Oeste, a incidência de AIDS tende à estabilização. No Norte e Nordeste, a tendência é de crescimento. Segundo critérios da Organização Mundial de Saúde (OMS), o Brasil tem uma epidemia concentrada, com taxa de prevalência da infecção pelo HIV de 0,6% na população de 15 a 49 anos (BRASIL, 2007).
Gráfico 1: Número de Casos de AIDS, segundo fonte de informação e ano de diagnóstico. Brasil, 1980-2006.
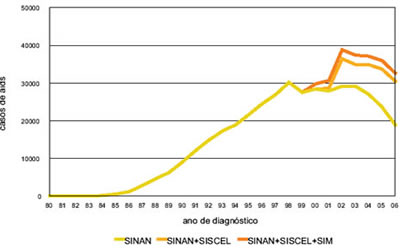
Fonte: (BRASIL, 2007)
O Boletim Epidemiológico 2007 trouxe, pela primeira vez, dados sobre a proporção de pessoas que continuaram vivendo com AIDS em até cinco anos após o diagnóstico. O estudo foi feito com base no número de pessoas identificadas com a doença em 2000. Os dados apontam que, cinco anos depois de diagnosticadas, 90% das pessoas com AIDS no Sudeste estavam vivas. Nas outras regiões, os percentuais foram de 78%, no Norte; 80%, no Centro Oeste; 81%, no Nordeste; e 82%, no Sul. A análise mostra, ainda, que a média de mortes dos indivíduos diagnosticados com AIDS no Brasil em até um ano após a descoberta da doença foi de 16,1%. Em números absolutos, o Brasil registrou 193 mil óbitos por AIDS, de 1980 a 2006 (BRASIL, 2007).
Na série histórica, foram identificados 314 mil casos de AIDS em homens e 160 mil em mulheres. Ao longo do tempo, a razão entre os sexos vem diminuindo de forma progressiva. Em 1985, havia 15 casos da doença em homens para 1 em mulher. Hoje, a relação é de 1,5 para 1. Na faixa etária de 13 a 19 anos, há inversão na razão de sexo, a partir de 1998. Em ambos os sexos, a maior parte dos casos se concentra na faixa etária de 25 a 49 anos. Porém, nos últimos anos, tem-se verificado aumento percentual de casos na população acima de 50 anos, em ambos os sexos (BRASIL, 2007).
Em 2006, foram registrados 526 casos em menores de 5 anos, representando taxa de incidência de 2,9 casos por 100 mil habitantes, mas esse número provavelmente está sub notificado. Considerando as regiões, a taxa de incidência é maior no Sul (6,1), seguido do Sudeste (4,4); Nordeste (3,1); Norte (2,7) e Centro Oeste (2,6) (BRASIL, 2007).
Quase 91% da população brasileira de 15 a 54 anos citam a relação sexual como forma de transmissão do HIV e 94% citam o uso de preservativo como forma de prevenção da infecção. O conhecimento é maior entre as pessoas de 25 a 39 anos, entre os mais escolarizados e entre as pessoas residentes nas regiões Sul e Sudeste. Os indicadores relacionados ao uso de preservativos mostram que aproximadamente 38% da população sexualmente ativa usam preservativo na última relação sexual, independentemente da parceria. Este número chega a 57% quando se consideram apenas os jovens de 15 a 24 anos. O uso de preservativos na última relação sexual com parceiro eventual foi de 67%. A proporção comparável em 1998 foi de 63,7% (BRASIL, 2007).
Até o junho de 2006, 77.639 casos de AIDS (17,9% do total do país) foram notificados na região Sul, sendo 19.495 casos em Santa Catarina. As maiores taxas de incidência ainda estão na região sul, porém com uma provável desaceleração do crescimento nos anos mais recentes (MANENTI, 2008).
Em 1986 foi registrado o primeiro caso de AIDS em Criciúma. Desde então, o crescimento da epidemia não foi constante, em 2002 foram registrados 82 casos, enquanto em 2005 diminuiu para 70 novos casos. Em 2006, manteve-se em 80 o número de notificações. No total de 21 anos (1986 à dezembro de 2007) de epidemia, o município de Criciúma acumulou 1.124 casos de AIDS. A faixa etária mais afetada é a de 20 a 34 anos, com 53,1 % dos casos. Até agosto de 2006 foram registradas 429 mortes por AIDS. Em 1997 a razão entre mortos e novos casos era de 1,7. A partir de 1998 essa razão decresceu chegando a 0,54 em 2002, e 0,35 no ano de 2005. Percebe-se que o numero de óbitos vem diminuindo (SINAN, 2008).
4.1.2 Organização, Estrutura e Genoma do Vírus
O agente etiológico da AIDS é membro da família Retroviridae, gênero Lentivirus (LORETE, 2005).
O HIV-1 é um vírus de formato esférico de aproximadamente 100nm de diâmetro contendo um nucleocapsídeo denso sob a forma de bastão. É um vírus envelopado, seu capsídeo é constituído de proteínas internas (LORETE, 2005), como as estruturais do core ou cerne viral (BISMARA, 2006), e nele está contido seu material genômico (RNA), além de enzimas necessárias para sua replicação (LORETE, 2005). As proteínas do envelope são codificadas pelo gene env, as proteínas do core são codificadas pelo gene gag e as proteínas com atividade enzimática são codificadas pelo gene pol (BISMARA, 2006) envolvido no desenvolvimento de resistência viral (AFANI et al., 2005).
A proteína da membrana externa do HIV (envelope-gp120) está ligada ao vírus pela glicoproteína da transmembrana (gp41). As proteínas p24 (kDa), p17 (kDa), p7 (kDa) são originadas de uma proteína de 55 kDa e formam as proteínas do nucleocapsídeo (BISMARA, 2006).
Figura 2: Estrutura do HIV.
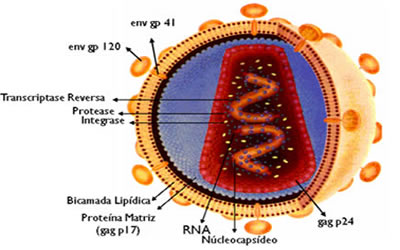
Fonte: (BISMARA, 2006)
Associadas ao complexo riboprotéico encontram-se as enzimas virais necessárias para replicação viral: transcriptase reversa, responsável pela retro-transcrição, a integrase, responsável pela integração do vírus ao genoma da célula hospedeira e a protease, responsável pelo processamento de precursores protéicos nas etapas finais da replicação (LORETE, 2005).
Envolvendo este complexo ribonucléico encontramos a proteína p24 responsável pela formação do capsídeo que constitui o core cilíndrico da partícula viral madura. No espaço entre o core e o envelope, encontramos a proteína p17 que por ser mistirilada na sua porção NH2-terminal se associa a parte interna do envelope estabilizando a estrutura do vírion (LORETE, 2005).
O genoma do HIV –1 codifica ainda seis outras proteínas acessórias (PEÇANHA et al., 2002), sendo quatro (nef, vif, vpr e vpu), que regulam a replicação do HIV através da produção de suas proteínas características (BISMARA, 2006), e duas (tat e rev) relacionadas com a regulação da expressão genética (PEÇANHA et al., 2002). A proteína nef tem a função de diminuir a expressão de CD4 e MHC (complexo principal de histocompatibilidade), bloquear apoptose, aumentar a infectividade viral, e alterar o estado de ativação da célula; a vif é um fator de infectividade (provavelmente atua na maturação da proteína do envelope); a vpr atua na replicação viral e ajuda na infecção de macrófagos; a vpu atua na liberação de partícula viral; a tat é um transativador da transcrição; e a rev é um transativador pós-transcricional (atuaria no processamento, transporte e tradução dos RNA mensageiros) (FOCACCIA & VERONESI, 2007).
Nas terminações 3’ e 5’, há longas seqüências repetidas denominadas long terminal repeats- LTRs (BISMARA, 2006) essenciais para a regulação da expressão dos genes virais, estando também envolvidos no processo de integração (LORETE, 2005).
4.1.3 Curso Clínico da Doença
Uma das principais características clínicas da AIDS é a imunossupressão, caracterizada pela destruição das células T CD4+. Estas células são responsáveis pela estimulação e manutenção da imunidade adquirida celular e humoral frente a vários microrganismos, além da ativação das células do sistema imune. Como conseqüências deste processo, podem ocorrer infecções oportunistas (infecções bacterianas, toxoplasmose, infecções fúngicas), caquexia, neoplasias (sarcoma de kaposi) e degeneração do Sistema Nervoso Central (SNC) (SILVEIRA, 2007).
Figura 3: Ciclo de Replicação Viral
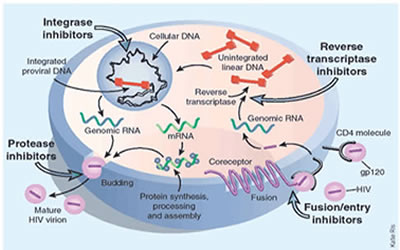
Fonte: (THE BIOLOGY PROJECT, 2000).
O primeiro passo para o inicio da infecção é a ligação da partícula viral a receptores específicos na superfície da célula alvo (PEÇANHA et al., 2002). A entrada ocorre através da fusão do vírus com a membrana da célula, sendo essa reação mediada pela gp 41 (FOCACCIA & VERONESI, 2007), onde, ocorre a ligação da proteína gp120 ao receptor CD4 (LOPES, 2007), expressa na superfície de células T e macrófagos (PEÇANHA et al., 2002). Alguns receptores de quimiocinas, principalmente CCR5 e CXCR4, são imprescindíveis para os passos subseqüentes da ligação do envelope viral à membrana plasmática. Somente após a ligação da gp120 a um desses receptores de quimiocinas é que ocorre uma alteração estrutural da gp120 expondo a gp41, que irá resultar na fusão da membrana celular com o envelope viral (LOPES, 2007).
A primeira fase de replicação do HIV, que inclui a entrada viral, a transcrição reversa e a integração do vírus no genoma do hospedeiro, é obtida por proteínas fornecidas pelo vírus. A segunda fase, que inclui a síntese e o processamento dos genomas virais do mRNA e das proteínas estruturais, utiliza a maquinaria celular do hospedeiro para a transcrição e para síntese protéica (STROHL et al., 2004).
A enzima do HIV, transcriptase reversa, catalisa a produção de uma cópia de DNA a partir do RNA do vírus HIV. A cópia de DNA de dupla hélice é então transportada ao núcleo celular onde uma segunda enzima do HIV, a integrase, catalisa a incorporação do DNA viral ao material genético do hospedeiro. A expressão subseqüente dos genes virais resulta na transcrição do RNA a partir do DNA do HIV e na tradução das proteínas virais. As proteínas virais recém-formadas são, no entanto, produzidas na forma de precursores de poliproteínas, longas unidades compostas de enzimas virais e proteínas estruturais ajuntadas. As poliproteínas e o RNA viral movem-se para a superfície da célula onde ficam incorporados aos novos vírus que brotam na membrana celular, levando parte da mesma com eles para formar a camada externa viral. Os vírus recém-formados seriam, no entanto, não infectantes sem a ação de uma terceira e essencial enzima do HIV, a protease, que processa as poliproteínas virais em proteínas e enzimas estruturais funcionais (SOUZA & ALMEIDA, 2003).
4.2 TRATAMENTO
O tratamento da infecção pelo HIV tem evoluído continuamente e mudando sensivelmente a historia natural da AIDS (FOCACCIA & VERONESI, 2007). Os inibidores da transcriptase reversa foram à primeira classe de fármacos, introduzidos como agente ARV para o tratamento do HIV e tem sido a base da terapia anti-HIV (LAVRA, 2006).Com o advento da TARV potente, as manifestações clinicas decorrentes da infecção pelo HIV tornaram-se menos freqüentes e houve melhora substancial do prognostico dos paciente com AIDS (MELO, 2007). A disponibilidade de diferentes classes de drogas ARV e o uso em combinação de três ou mais delas, transformou o tratamento dos indivíduos com AIDS, de tal modo que a morbidade e mortalidade declinaram entre 60% e 80% (DOMINGOS, 2006).
O conhecimento da dinâmica viral e o surgimento de métodos laboratoriais capazes de mensurar a quantidade de vírus circulante no plasma (carga viral) tornaram possível a monitorização confiável e objetiva da evolução e do tratamento da infecção pelo HIV (FOCACCIA & VERONESI, 2007). O ciclo de replicação do HIV apresenta diversos eventos exclusivamente relacionados a componentes virais, que podem se utilizados como alvos para intervenção quimioterápica (PEÇANHA et al., 2002).
Atualmente para o tratamento do HIV dispõe-se das seguintes classes de ARV: inibidores da transcriptase reversa análogos de nucleosídeos (ITRNs), inibidores da transcriptase reversa não análogos de nucleosídeos (ITRNNs), inibidores da protease (IPs), inibidores da integrase, antagonistas de CCR5 e inibidores de fusão (IsF) (HUGHES et al., 2008).
4.2.1 Inibidores da transcriptase reversa análogos de nucleosídeos (ITRNs)
Os Inibidores da transcriptase reversa análogos de nucleosídeos são pró-drogas análogas de dideoxinucleosídeos (LAVRA, 2006), utilizadas pela transcriptase reversa como substrato para síntese de DNA (GARFORTH et al., 2008). Os inibidores da transcriptase reversa impedem a produção da cópia de DNA a partir do RNA, através da inibição competitiva do Desoxinucleotideo trifosfato fisiológico, impedindo, assim, a extensão da fita (STROHL et al., 2004; MANENTI, 2008).
Para que tenham atividade antiviral contra a enzima transcriptase reversa, necessitam ser fosforilados pelo organismo pelas enzimas celulares chamadas de quinases (SOUZA & ALMEIDA, 2003), onde o metabólito, trifosfato, interrompe, competitivamente, a replicação viral (LAVRA, 2006).
É importante destacar a utilidade de fármacos análogos de nucleosídeos como terapêutica de primeira linha contra a infecção pelo HIV-1 (GARFORTH et al., 2008). Atualmente, no mercado, existem oito medicamentos nucleosídeo-nucleotídeo capazes de inibir a enzima transcriptase reversa: zidovudina (AZT), Estavudina (d4T), zalcitabina (ddC), lamivudina (3TC), didanosina (ddI), abacavir (ABC), Emtricitabina (FTC) e Tenofovir disoproxil fumarato (TDF) (SOUZA & ALMEIDA, 2003).
Zidovudina – AZT
Zidovudina é um potente agente antiviral utilizado no tratamento da AIDS (RAVI et al., 2008). A AZT é um análogo da timidina. Pode prolongar a vida dos indivíduos infectados por HIV e na demência moderada associada ao HIV (RANG & DALE, 2005).
Recentemente foi demonstrado que pacientes com decréscimo na função cognitiva apresentaram melhora em seu QI após a TARV ser alterada para incluir zidovudina (HAZRA et al., 2007). O AZT Pode reduzir a transmissão transplacentária, quando administradas pela gestante, em mais de 20% (RANG & DALE, 2005).
A posologia recomendada geralmente é de 300 mg duas vezes ao dia (LAVRA, 2006) devido a sua meia-vida curta, porém esta posologia apresenta muitas desvantagens tais como efeitos colaterais adversos, devido ao acúmulo de droga em multi-dose terapêutica, além do alto custo (RAVI et al., 2008). Estudos demonstram o curto prazo de tolerabilidade em mulheres, em indivíduos com baixo índice de massa corporal (IMC), e principalmente, o risco de anemia em pacientes com doença avançada (ISAAKIDIS et al., 2008). Portanto, o ideal seriam doses de 600 mg uma vez ao dia para que se possam minimizar alguns destes problemas providos do acumulo de droga em multi-dose. (RAVI et al., 2008).
A biodisponibilidade é de 60 – 80%, e a concentração plasmática máxima é alcançada em 30 minutos. A meia-vida é de 1 hora, enquanto a meia-vida intracelular do trifosfato ativo é de 3 horas. A concentração no liquido cefalorraquidiano (LCR) atinge 65% dos níveis plasmáticos. A maior parte do fármaco é metabolizada a glicuronídio inativo no fígado, e apenas 20% da forma ativa são excretados na urina. O AZT é menos ativado na forma trifosfato, portanto torna-se mais vulnerável a resistência viral (RANG & DALE, 2005).
Nas duas primeiras semanas de terapia são comuns as queixas de náuseas e cefaléias, que costumam desaparecer após este período. Podem ainda surgir vômitos, febre, exantema, parestesias e mialgias. A toxicidade medular, exteriorizada por anemia e neutropenia, ocorre principalmente em pacientes com formas avançadas da doença. Um efeito colateral raro, mas que pode apresentar significativa gravidade, é a acidose lática. Hepatotoxicidades, hepatomegalia e esteatose hepática também ocorrem com baixa freqüência (LOPES 2007). Alguns estudos têm documentado que o tratamento de pacientes HIV positivos com AZT está associado ao aumento da aderência e formação de biofilmes de C. albicans oral (AHIDJO et al., 2008).
Estavudina - d4T
Após fosforilação intracelular, assume a forma de trifosfato de estavudina, competindo com o nucleotídeo natural timidina na formação de acido nucléico resultando na interrupção da formação da cadeia. É rapidamente absorvida após a administração via oral, com biodisponibilidade de cerca de 86% (LOPES, 2007).Tem meia-vida plasmática de 1 hora (RANG & DALE, 2005) e meia-vida intracelular prolongada, cerca de 3,5 horas. Atinge concentrações no LCR de cerca de 30 a 40% equivalentes a concentração sérica. Atravessa a barreira placentária (LOPES, 2007).
Não há necessidade de jejum para sua administração e as doses recomendadas para adultos são de 40 mg em intervalos de doze horas para pacientes com peso igual ou superior a 60 kg e 30 mg em intervalos de 12 horas para pacientes abaixo de 60 kg. É encontrado na forma de comprimidos de 15, 20, 30 e 40 mg (LOPES, 2007).
A maior parte é eliminada pelos rins por secreção tubular ativa (RANG & DALE, 2005). d4T é conhecido por ser a causa de uma boa parcela dos efeitos colaterais sofridos por pacientes em TARV. Sintomas como neuropatia periférica, acidose láctica, e lipodistrofia e lipoatrofia são atribuídos a todos os d4T (ROSEN et al., 2008). Também pode ocorrer hepatite, sendo esta, freqüentemente oligossintomática ou assintomática, detectada por aumento dos níveis séricos de enzimas hepáticas (LOPES, 2007). Outros efeitos adversos incluem dor articular e pancreatite (RANG & DALE, 2005). As taxas tóxicas de d4T são maiores em Africanos do que os encontrados em ensaios clínicos em países industrializados, relatado em estudos de coorte (ROSEN et al., 2008).
Zalcitabina – ddC
A zalcitabina é análoga da citosina que possui atividade anti-HIV-1 sinérgica com uma variedade de agentes anti-retrovirais contra cepas de HIV-1 tanto sensíveis quanto resistentes ao AZT (KATZUNG, 2006). É ativada na célula T por uma via de fosforilação diferente daquela da AZT (RANG & DALE, 2005). Possui meia-vida intracelular relativamente longa de 10 horas (Apesar de sua meia-vida de eliminação de 2 horas) e alta biodisponibilidade oral (> 80%). Todavia, os níveis plasmáticos diminuem em 25-39% quando o fármaco é administrado com alimento ou com antiácidos. A ligação às proteínas plasmáticas é baixa (< 4%). As concentrações no LCR são aproximadamente 20% daquelas obtidas no plasma (KATZUNG, 2006).
O efeito indesejável mais importante consiste em neuropatia relacionada com a dose (que pode aumentar durante varias semanas após a interrupção do fármaco) (RANG & DALE, 2005). O potencial de neuropatia periférica constitui uma contra-indicação relativa para o uso da zalcitabina com outros fármacos passiveis de causar neuropatia, incluindo estavudina, didanosina e isoniazida (KATZUNG, 2006). Outros efeitos indesejáveis incluem distúrbios gastrintestinais, cefaléia, úlceras bucais, alterações ungueais, edema dos membros inferiores e mal-estar geral. Ocorrem exantemas, que podem desaparecer espontaneamente (RANG & DALE, 2005). A pancreatite ocorre menos freqüentemente do que com o uso da didanosina, porem a co-administração de outros fármacos que provocam pancreatite pode aumentar a freqüência desse efeito adverso (KATZUNG, 2006).
As interações medicamentosas potenciais incluem aumento da ASC (área sob a curva) da zalcitabina quando administrada em associação com probenecida ou cimetidina e diminuição da biodisponibilidade quando a zalcitabina é administrada concomitantemente com antiácidos ou metoclopramida. A 3TC inibe a fosforilação da zalcitabina in vitro, interferindo potencialmente na sua eficácia (KATZUNG, 2006).
Embora se tenha descrito uma variedade de mutações associadas à resistência in vivo a zalcitabina (por exemplo, T69D, K65R, M186V), a resistência fenotípica parece ser rara, particularmente em esquemas de associação (KATZUNG, 2006).
Lamivudina - 3TC
É um nucleosídeo sintético que, após fosforilação, trona-se um análogo a nucleotídeos naturais, inibindo a ação da transcriptase reversa e interrompendo a cadeia de formação de ácidos nucléicos (LOPES, 2007). Análogo da pirimidina é o único fármaco aprovado pelo FDA (Food and Drug Administration) que tem atividade contra HIV-1, HIV-2 e hepatite B (LAVRA, 2006).
É bem absorvido após ingestão oral, com biodisponibilidade de 80 a 86%. Sua meia-vida sérica é de 3 a 6 horas, ao passo que a intracelular é de 12 horas. Atinge concentrações baixas no liquido cefalorraquidiano, cerca de 10% da concentração no soro (LOPES, 2007). A maior parte é excretada na forma inalterada na urina (RANG & DALE, 2005).
A terapia com lamivudina leva rapidamente a seleção, tanto in vitro quanto in vivo, de mutantes de HIV M184V-resistentes, conferindo um alto nível de resistência a lamivudina e uma redução da sensibilidade ao abacavir, à didanosina e a zalcitabina. A mutação M184V pode restaurar a sensibilidade fenotípica a zidovudina, indicando que esse dois fármacos administrados em esquemas de combinação podem ser particularmente benéficos (KATZUNG, 2006).
Efeitos colaterais, como neuropatia periférica, pancreatite, cefaléia, tonturas e insônia, são raras. Praticamente não apresenta toxicidade hematológica, com raros relatos de anemia ou neutropenia seguramente relacionados ao seu uso. A dose habitualmente indicada para adultos é de 150 mg a cada doze horas, que requer ajuste em pacientes com insuficiência renal. É encontrada na formulação de comprimidos contendo 150 mg de lamivudina isolada ou associada a 300 mg de zidovudina. Recentemente também se tornou disponível a formulação de comprimidos contendo 150 mg de lamivudina, 300 mg de zidovudina e 300 mg de abacavir (LOPES, 2007).
Didanosina – DDI
A didanosina ou DDI é um análogo da purina com atividade ARV contra os vírus HIV-1 e HIV-2 incluindo as cepas resistentes à zidovudina (AZT). Intracelularmente é convertida pela enzima 5-nucleotidase em dd-IMP (dideoxytinosina 5 monofosfato) que por fim é metabolizada em sua forma ativa dd-ATP (2-3 dideoxyadenosina trifosfato), funcionando como um competidor do substrato dATP da enzima transcriptase reversa viral (inibidor competitivo) (MURALHA et al., 2001).
É administrada por via oral, sofre rápida absorção em meio básico, o que obriga sua administração em comprimidos formulados com antiácidos, longe das refeições (LOPES, 2007), e é ativamente secretada pelos túbulos renais (RANG & DALE, 2005). As doses recomendadas são de 200 mg a cada 12 horas ou uma tomada de 400 mg, em indivíduos com peso acima de 60 kg (LOPES, 2007).
Sua biodisponibilidade é de 25 a 40%, atingindo picos séricos cerca de 30 minutos após ingestão. Sua meia-vida sérica é de 60 a 90 minutos, mas sua meia-vida intracelular é muito mais longa, entre 8 e 24 horas, permitindo a administração em intervalos de 12 ou até 24 horas. Atravessa a barreira hemato-encefálica, com níveis no LCR 19 a 21% dos observados no plasma em adultos, e mais elevados em crianças. Atravessa a barreira placentária e é removida por hemodiálise (LOPES, 2007).
A neuropatia periférica e a pancreatite são considerados os principais efeitos colaterais dose dependentes da didanosina, ocorrendo geralmente nos primeiros 3 a 6 meses de uso. Casos de retinopatia tóxica em crianças HIV positivas são relatados associados ao uso de altas doses de DDI, porém raros casos são relatados em adultos (MURALHA et al., 2001).
Abacavir – ABC
O abacavir é um análogo da guanosina (RANG & DALE, 2005), com atividade contra o HIV, disponível para utilização em combinação com outros agentes anti-retrovirais que tem demonstrado, eficácia, poucas interações medicamentosas, e uma favorável, a longo prazo, toxicidade da substância (MALLAL et al., 2008). É bem absorvido após administração por via oral, independente do estado de alimentação, proporcionando biodisponibilidade de 80 a 95 %. Sua meia-vida sérica é de cerca de 0,9 a 1,7 hora e sua ligação a proteínas plasmáticas ao redor de 50%. Tem boa penetração no liquido cefalorraquidiano, similar a AZT (LOPES, 2007). É metabolizado no fígado a compostos inativos. (RANG & DALE, 2005).
A dose empregada em adultos é de 300 mg por via oral a cada 12 horas. É disponível em comprimidos de 300 mg ou em associação com 150 mg de lamivudina e 300 mg de zidovudina (LOPES, 2007).
O efeito adverso mais importante do abacavir, que limita a sua utilização na terapia e fazendo com que se mantenha um alto grau de vigilância clínica, é uma reação imunológica de hipersensibilidade mediada afetando 5 a 8% dos pacientes durante as primeiras 6 semanas de tratamento. Os sintomas de uma reação de hipersensibilidade ao abacavir incluem combinações de febre, erupção cutânea, sintomas constitucionais, sintomas do trato gastrointestinal, sintomas respiratórios e tornam-se mais grave com a administração contínua. A imediata e definitiva interrupção do tratamento é obrigatória, resultando em uma rápida reversão dos sintomas. Os sintomas da reação de hipersensibilidade ao abacavir são inespecíficos e podem ser difíceis de distinguir das infecções concomitantes, reação a outros medicamentos, ou doença inflamatória (MALLAL et al., 2008). Estudos sugerem um inesperado aumento do risco de infarto do miocárdio associado à terapia com abacavir (CUNTRELL et al., 2008).
Emtricitabina - FTC
Emtricitabina, FTC fluorado é um derivado da lamivudina (3TC), um análogo da deoxicitidina, que é ativo contra HIV-1, HIV-2 e vírus da hepatite B. Foi aprovado pela FDA para uso desde 2003 e é atualmente recomendado como parte de um regime terapêutico inicial preferível. Em comparação com 3TC, FTC tem uma meia-vida longa (MASHO et al., 2007) (cerca de 10 horas), o que permite seu uso em dose única diária de 200 mg (LOPES, 2007), maior biodisponibilidade oral, e ligeiramente maior potência in vitro, embora o significado clínico deste permaneça incerto (MASHO et al., 2007).
Os estudos indicam que a FTC é eficaz em combinação com outros ARV, como o tenofovir, na redução da carga viral em ambos os tratamentos de pacientes experientes (em uso de ARV) e virgens de tratamento. FTC pode ser tomada sem se preocupar com o consumo alimentar e é totalmente eliminada através dos rins, por isso é necessário ajuste da dose em caso de insuficiência renal (MASHO et al., 2007) e evitada em pacientes com clearance de creatinina abaixo de 30 mL/minuto (LOPES, 2007).
Potenciais interações medicamentosas e de toxicidade, incluindo a toxicidade mitocondrial e dislipidemias, são quase inexistentes. Os efeitos secundários mais comuns incluem dor de cabeça, insônia, diarréia, náuseas, vômitos, erupções e cutâneas. FTC pode causar hiperpigmentação da pele das palmas das mãos e nas solas de pacientes Africanos e afro-americanos (MASHO et al., 2007). Pacientes com hepatite B crônica em uso de emtricitabina podem apresentar reagudização da hepatite B logo após a sua suspensão (LOPES, 2007).
Tenofovir disoproxil fumarato - TDF
Aprovado pela FDA em 2001, TDF (um éster pró-droga do tenofovir) é hidrolisado a tenofovir intracelular e fosforilado ao metabolito ativo, tenofovir difosfato. O tenofovir é um análogo nucleotídeo da deoxiadenosina monofosfato, com atividade contra o HIV-1, HIV-2 e vírus da hepatite B (HBV). Devido à sua meia-vida longa (17 horas), é administrada uma vez por dia com outras drogas anti-retrovirais (MASHO et al., 2007). O TDF tem sido proposto como droga candidata a prevenção ao HIV perinatal (CHI et al., 2008).
Diversos ensaios clínicos indicaram que TDF é muito potente, em ambos os tratamentos de pacientes ingênuos e experientes com HIV e reduziram significativamente a carga viral. Foi igualmente demonstrado que é uma alternativa eficaz como agente antiviral durante a falha terapêutica ou toxicidade a droga entre os doentes com experiência de tratamento. Ela pode ser tomada sem que se consuma alimento, mas é absorvido 39% quando tomado com uma refeição gordurosa comparado a 25% quando administrada antes de uma refeição (jejum) (MASHO et al., 2007).
TDF é excretada inalterada pelos rins, assim, toxicidade tubular renal é um efeito colateral importante, mas pouco freqüente. É recomendado ajuste posológico na insuficiência renal (MASHO et al., 2007), e observações freqüentes da função renal após o início do tratamento com tenofovir devido à possibilidade de aparecimento tardio de insuficiência renal (KAPITSINOU & ANSARI, 2008). Uma importante preocupação da segurança para seu uso em adultos é o potencial efeito adverso sobre o metabolismo ósseo, demonstrado tanto em animais quanto em humanos (GAFNI et al., 2008). TDF não é metabolizada pelas enzimas do citocromo P450 (CYP450), por tanto, não existe potencial de interação com fármacos metabolizados por estas enzimas. TDF induz pouca ou nenhuma toxicidade mitocondrial ou dislipidemia. Outras reações adversas ocasionais incluem náuseas, diarréia, vômitos, erupção cutânea, e flatulência (MASHO et al., 2007).
O surgimento de resistência viral pode ocorrer rapidamente, tal como quando TDF é utilizado em associação com didanosina e efavirenz ou com abacavir e lamivudina. O segredo das mutações de resistência contra a TDF é uma interrupção da lisina-arginina na posição 65, no gene da transcriptase reversa (ou seja, K65R), que exige apenas um nucleotídeo para mudar a base (WYL et al., 2008).
4.2.2 Inibidores da transcriptase reversa não análogos de nucleosídeos (ITRNNs)
Ligam-se de modo não-competitivo e reversível a transcriptase reversa, alterando assim sua função. Suas principais vantagens são a ausência de efeitos sobre os elementos formadores do sangue do hospedeiro e a ausência de resistência cruzada com os inibidores da transcriptase reversa análogos de nucleosídeos (STROHL et al., 2004). Os ITRNNs são metabolizados principalmente pela via do CYP450 enzimática (BIANCO, 2007).
Esta classe de fármacos possui maior eficácia contra HIV-1 do que contra HIV-2. É ativo em seu estado nativo e não requer fosforilação. A forma ideal de utilização destes fármacos é na terapia combinada com análogos de nucleotídeos e inibidores de protease, onde a atividade sinérgica deles é mais potente (LAVRA, 2006).
Para tratamento com não análogos de nucleosídeos, capazes de inibir a enzima transcriptase reversa, existem no mercado três medicamentos: nevirapina (NVP), efavirenz (EFZ) e delavirdina (SOUZA & ALMEIDA, 2003).
Nevirapina – NVP
Foi o primeiro ITRNN, introduzido em 1996, indicado em pacientes que tem demonstrado falência imunológica e virológica, e deve ser usado em combinação com análogos de nucleosídeos (LAVRA, 2006).
Liga-se diretamente a TR, bloqueando a atividade de polimerase da enzima por incapacitar seu sitio catalítico. Bem absorvido por via oral, não sofre interferência por ingestão de alimentos ou antiácidos (LOPES, 2007). Apresenta biodisponibilidade > 90%, e os níveis alcançados no LCR correspondem a 45% dos níveis plasmáticos (RANG & DALE, 2005). Possui meia-vida plasmática superior a 24 horas, o que permite seu uso em dose única diária, embora a recomendação mais comum seja de administração duas vezes ao dia (LOPES, 2007). Atinge concentração plasmática 3 horas após uma dose única de 200 mg (KONDO et al., 2008).
O uso recomendável é de 200 mg diariamente nas primeiras duas semanas, seguindo pelo aumento da dose para 200 mg duas vezes ao dia a partir da terceira semana de tratamento. É uma das melhores opções para tratamento da infecção pelo HIV em pacientes com diagnóstico de complexo demencial da AIDS (LOPES, 2007).
O fármaco é metabolizado no fígado pelo sistema enzimático CYP450. Curiosamente, a nevirapina induz seu próprio metabolismo, interferindo nas concentrações plasmáticas nas primeiras duas e quatro semanas de tratamento (LOPES, 2007). Seu metabólito é excretado na urina (RANG & DALE, 2005). O perfil farmacocinético favorável da nevirapina permite dosagem simplificada e regime pouco oneroso para prevenir a transmissão perinatal, útil especialmente em países em desenvolvimento. A nevirapina tem sido amplamente utilizada no Brasil como parte do esquema tríplice ARV durante a gestação. (KONDO et al., 2008).
A erupção cutânea é o efeito colateral mais comum da nevirapina e sua incidência ocorre entre 15 – 20% dos indivíduos (MUNDERI et al., 2008). Geralmente, aparece nas primeiras quatro semanas e pode resultar no desenvolvimento da síndrome de Stevens-Johnson (erupção bolhosa que acomete o tegumento e as mucosas) ou necrose epidérmica tóxica (KONDO et al., 2008). Nesses casos, a droga deve ser interrompida e seu uso futuro evitado. Aumento das transaminases hepáticas também é freqüente, recomendando cautela no uso em pacientes que são cronicamente infectados pelos vírus das hepatite B e C, em mulheres com contagem de LT CD4+ acima de 250 cels/mL e em homens com contagem de LT CD4+ acima de 400 cels/mL (LOPES, 2007).
Efavirenz - EFZ
O EFZ é um fármaco ARV de primeira linha para a infecção pelo HIV (QUEREDA et al., 2008). É bem absorvido por via oral, sem sofrer interferência significativa dos alimentos. Sua longa meia-vida plasmática (>24 horas) permite seu uso uma vez ao dia. Tem boa penetração no SNC. Após a sua administração, 99% ligam-se a albumina plasmática, e a concentração do LCR corresponde ~1% das concentrações plasmáticas. O fármaco é inativado no fígado (RANG & DALE, 2005).
Metabolizado pelo sistema enzimático CYP450, sofre e exerce interferência no nível sérico de vários outros medicamentos, incluindo ritonavir, indivavir, SQV, claritromicina, cisaprida etc. Recomenda-se a verificação da possibilidade de interação significativa sempre que for utilizado medicamento que interage com o mesmo sistema enzimático hepático no seu metabolismo. Encontram-se na apresentação de comprimidos com 200 e 600 mg e é utilizado na dose de 600 mg uma vez ao dia (LOPES, 2007).
A reação adversa mais comum envolve efeitos colaterais neuropsiquiátricos, principalmente humor e distúrbios do sono, tanto a curto quanto a longo prazo (QUEREDA et al., 2008), exteriorizada por queixas de tonturas, dificuldades de concentração, cefaléia e alteração dos sonhos. Esta ultima e curiosa queixa consiste em vivacidade incomum dos sonhos e freqüente lembrança após o despertar. É importante alertar os pacientes sobre tais efeitos antes do inicio do uso do EFZ e recomendar a ingestão antes de dormir, o que ameniza o desconforto durante o dia. Esses sintomas tendem a regredir após as primeiras semanas de uso. Podem também ocorrer exantema e sintomas gastrintestinais leves (LOPES, 2007).
Um estudo demonstrou uma correlação de hipersusceptibilidade fenotípica entre o EFZ e diferentes números de mutações da TR em análises variadas, incluindo T215Y / F, D67N, H208Y, K103R, e V179D. Análises posteriores mostraram uma associação entre as várias mutações e hipersusceptibilidade fenotípica, e a escalonada regressão logística, constatou que três códons, 118, 208, e 215, permaneceram preditivas independente da hipersusceptibilidade do EFZ (DEMETER et al., 2008).
EFZ tem sido ligada a defeitos congênitos em animais e defeitos de tubo neural em humanos, no caso de exposição durante o primeiro trimestre da gravidez. Embora a prevalência de alterações em seres humanos relatados na literatura é baixa, a recomendação consensual é de que tratamento com EFZ deve ser evitado durante o primeiro trimestre da gravidez (SCHNEIDER et al., 2008).
Delavirdina
A Delavirdina possui biodisponibilidade oral de cerca de 85%, que é reduzido na presença de antiácidos. O fármaco liga-se extensamente (cerca de 98%) as proteínas plasmáticas. Os níveis alcançados no LCR correspondem, em media, a apenas 0,4% das concentrações plasmáticas, representando cerca de 20% da fração não-ligada as proteínas plasmáticas. Deve-se ter cautela ao administrar-se delavirdina aos pacientes com insuficiência hepática, visto que a experiência clinica nessa situação é limitada (KATZUNG, 2006).
Ocorre erupção cutânea em cerca de 18% dos pacientes tratados com delavirdina; tipicamente, aparece no primeiro mês de terapia e não impede uma nova exposição ao fármaco. Entretanto foi relatada a ocorrência de exantema grave, como eritema multiforme e síndrome de Stevens-Johnson. Outros efeitos adversos podem incluir cefaléia, fadiga, náuseas, diarréia e níveis séricos aumentados de aminotransferases. Foi constatado ser a delavirdina teratogênica em ratos, causando defeitos do septo ventricular e outras más formações com exposição ao fármaco que não diferem daquelas obtidas em seres humanos. Por conseguinte, deve-se evitar a gravidez durante o uso de delavirdina (KATZUNG, 2006).
A delavirdina é extremamente metabolizada a metabólitos inativos pelas enzimas CYP3A e CYP2D6. Todavia, o fármaco também inibe a CYP3A e, portanto, inibe o seu próprio metabolismo. Alem de suas interações com outros agentes ARV, a delavirdina resulta em níveis aumentados de numerosos fármacos. Deve-se considerar uma redução da dose de indinavir e SQV se esses fármacos forem administrados concomitantemente com delavirdina. As concentrações plasmáticas de delavirdina diminuem na presença de antiácidos, fenitoína, fenobarbital, carbamazepina, rifabutina e rifampicina; observa-se um aumento das concentrações durante a co-administração com claritromicina, fluoxetina, dexametasona e cetoconazol (KATZUNG, 2006).
4.2.3 Inibidores da Protease (IPs)
Os produtos dos genes gag e pol são traduzidos inicialmente em grandes poliproteínas precursoras, que devem ser clivadas pela protease viral para produzir as proteínas maduras. Os inibidores da protease interferem no processamento das poliproteínas no vírion em brotamento e resultam em partículas não-infecciosas. O maior problema encontrado para este fármaco, é que eles causam lipodistrofia (redistribuição de gordura, de modo que os membros tornam-se magros e a gordura é depositada ao longo do abdômen e do dorso superior) e hiperglicemia (STROHL et al., 2004).
São dez os medicamentos capazes de inibir a enzima protease existentes no mercado: saquinavir (SQV), ritonavir (RTV), indinavir (IDV), lopinavir (LPV/r), nelfinavir, amprenavir, atazanavir (ATV), fosamprenavir (FPV), tipranavir e darunavir (SOUZA & ALMEIDA, 2003).
Saquinavir - SQV
Foi o primeiro IP disponibilizado para uso. Em sua formulação inicial foi produzido em cápsulas rígidas, com baixa absorção e biodisponibilidade de somente 4%. Recentemente, outra formulação, em cápsulas gelatinosas, melhorou significativamente esse problema. É melhor absorvido quando ingerido com alimentos, especialmente os ricos em gordura (LOPES, 2007). Possui um grande volume de distribuição, porém 98% de uma dose ligam-se a proteínas. A penetração do fármaco no liquido LCR é insignificante. A meia-vida de eliminação é de 12 horas. A excreção ocorre primariamente nas fezes (KATZUNG, 2006).
Apresenta interação com vários medicamentos, incluindo diversos ARV, além de outros também metabolizados preferencialmente pelo complexo enzimático CYP450. O SQV sofre extenso metabolismo de primeira passagem pela CYP450 e atua tanto como inibidor quanto como substrato da CYP450. A co-administração com o inibidor da CYP450, ritonavir, foi adotado pelos médicos, visto que a inibição do metabolismo de primeira passagem do SQV pelo ritonavir pode resultar em níveis mais elevados, e, portanto, mais eficazes, de SQV (KATZUNG, 2006). As reações adversas incluem diarréia, náuseas, dor abdominal e epigástrica, que se acentuam com o uso de formulação em cápsulas gelatinosas, provavelmente refletindo a maior absorção. Pode produzir elevação de triglicerídeos, enzimas hepáticas e creatina- fosfoquinase (LOPES, 2007). É preciso monitorizar as provas de função hepática quando se administra SQV concomitantemente com delavirdina (KATZUNG, 2006).
É usado em associação com ritonavir, na dose de 400 mg de cada droga a cada doze horas, ou na dose de 1.000 mg de SQV com 100 mg de ritonavir, a cada doze horas. Disponível em comprimidos de 200 mg (LOPES, 2007).
As mutações críticas mais comuns são L90M e G48V, conferindo diminuição de cerca de 10 vezes na suscetibilidade ao fármaco (KATZUNG, 2006).
Ritonavir - RTV
O Ritonavir é um sal do ácido paratoluenossulfônico, com ampla distribuição tecidual, metabolizado pelo fígado e por ele excretado, embora uma pequena quantidade, inferior a 5%, seja eliminada pelos rins. É um inibidor de protease viral com especificidade para o vírus HIV-1, utilizado com freqüência nos esquemas de ARV combinados para gestantes contaminadas (CARVALHO et al., 2007).
Bem absorvido por via oral, possui meia-vida plasmática de 3,2 horas (LOPES, 2007). Tem alta biodisponibilidade (cerca de 75%), que aumenta quando o fármaco é ingerido com alimento (KATZUNG, 2006). É um potente inibidor e é metabolizado pela enzima 3A4 do complexo enzimático CYP450. Isso faz com que seu uso se resulte em alterações significativa dos níveis séricos de varias outras drogas metabolizadas por essa enzima. Essa característica é de extrema importância quando são considerados associações de drogas, tanto se beneficiando dessa propriedade do RTV como para evitar efeitos colaterais (LOPES, 2007).
Mais da metade dos pacientes apresenta efeitos colaterais gastrointestinais, como náuseas, vômitos e diarréia, quando RTV é usado em sua dose plena, de 600 mg a cada doze horas. Também esta associada à anorexia, dor abdominal, fraqueza, parestesia periférica e perioral, alterações do paladar e cefaléia. Igualmente são descritas alterações laboratoriais como elevação do colesterol, triglicérides enzima hepáticas e creatina- fosfoquinase (LOPES, 2007).
Alguns especialistas recomendam iniciar o tratamento com 300 mg a cada doze horas, aumentando progressivamente por uma ou duas semana até a dose plena de 600 mg a cada dose horas. Contudo, o uso do RTV tem crescido muito como adjuvante a outros ARV, atuando no aumento do pico da concentração plasmática e prolongamento da meia-vida dessas outras drogas. O RTV é disponível em comprimidos de 100 mg (LOPES, 2007).
A redução na atividade anti-retrovíral de RTV está essencialmente associada às mutações V28A/F/T/S e 184V da protease. A acumulação de outras mutações no gene da protease (incluindo nas posições 20, 33, 36, 46, 54 71 e 90) pode também contribuir para resistência ao ritonavir (KATZUNG, 2006).
Indinavir – IDV
Quando utilizado com único IP, necessita de meio ácido para ser bem absorvido, sofrendo interferência ao ser ingerido com alimentos, especialmente com alto teor de gorduras. Isso faz com que deva ser administrado longe das refeições, uma hora antes ou duas horas depois com alimentos leves. Disponível em comprimidos de 400 mg, apresenta meia-vida plasmática de 1,8 hora e deve ser usado na dose de 800 mg, em intervalos de oito horas (LOPES, 2007). A biodisponibilidade oral é de cerca de 65%, e aproximadamente 60% do fármaco se ligam às proteínas plasmáticas. O IDV é o que exibe maior penetração no LCR entre os IPs, atingindo 76% dos níveis séricos (KATZUNG, 2006). Aproximadamente 20% de uma dose oral de IDV é excretada em sua forma inalterada na urina (EIRA, 2004).
Igualmente metabolizado pelo complexo CYP450, apresenta interações com diversas drogas. A principal delas é com o ritonavir. Quando utilizados simultaneamente, ambos IP sofrem elevação dos níveis séricos. Portanto, uma alternativa ao uso isolado é combinar 800 mg de IDV com 100 ou 200 mg de RTV a cada doze horas. No primeiro caso, a associação é empregada em substituição a dose de 800 mg a cada oito horas; no segundo, a associação é utilizada principalmente no tratamento de resgate de pacientes que experimentaram falecia ao esquema ARV inicial. Outras interações incluem nevirapina, efavirenz, delavirdina, rifampicina, fluconazol, quetoconazol, claritromicina, alguns anti- histamínicos, ergotaminas, cisaprida e vários benzodiazepínicos. Apresenta antagonismo ARV com o SQV, o que contra-indica essa associação (LOPES, 2007).
Depois de metabolizado pelo fígado, é eliminado pelo rim, podendo precipitar em cristais. Esses cristais possibilitam o desenvolvimento de litíase renal, minimizada pela ingestão de mais de 1,5 L de líquidos todos os dias. A freqüência de litíase renal aumenta quando o IDV é utilizado em associação com o RTV, especialmente na combinação 800 mg/200 mg (LOPES, 2007). Seus principais efeitos adversos são a toxicidade renal, evidenciada pela nefro-litíase, e a dislipidemia, hipertensão e resistência periférica a insulina, que freqüentemente requerem intervenções terapêuticas. Também pode causar aumento das bilirrubinas no plasma (que não costuma ter conseqüências maiores) (FALCI et al., 2006). Pode ainda causar sintomas gastrintestinais, como náuseas, vômitos, dor epigástrica, além de insônia, xerostomia e xerodermia. Também pode causar alterações semelhantes as observada na terapia com ácido retinóico, incluindo paroníquia, que ocorrem em 4 a 5% dos casos, além da xerodermia e queilite. Tais manifestações são reversíveis com a suspensão da droga. (LOPES, 2007).
A resistência pode estar associada a múltiplas mutações, e o número de alterações de códons (tipicamente substituições) tende a prever o nível de resistência fenotípica. As mutações nas posições 46 e 82 do gene da protease parecem prever de modo mais consistente uma resistência fenotípica. A resistência ao IDV está associada a uma perda de sensibilidade ao RTV (KATZUNG, 2006).
Lopinavir – LPV/r
Vem co-formulado com RTV, fazendo com que consiga atingir níveis plasmáticos mais prolongados e duráveis pela inibição da isoenzima 3A4 do sistema enzimático CYP450. Sua biodisponibilidade absoluta, após ingestão oral em humanos ainda não foi estabelecida. Sua concentração plasmática máxima após ingestão de 400 mg de LPV/r e 100 mg de RTV é elevada, alcançando entre 2,1 e 8,1 horas. Sua absorção aumenta quando é ingerido com alimentos gordurosos. Sem a associação com o RTV, o LPV/r é rapidamente metabolizado. Já com o ritonavir, ocorre grande elevação dos níveis plasmáticos, dezenas de vezes superiores a concentração necessária para inibir o vírus selvagem (que não apresenta mutações genéticas que conferem resistência). Sua meia-vida plasmática é de cinco a seis horas, sendo eliminado pela urina e fezes (LOPES, 2007).
Ainda não são disponíveis recomendações para o ajuste da dose em pacientes com insuficiência renal ou hepática. Os efeitos colaterais gastrintestinais são os mais comuns, com náuseas, vômitos, flatulência e fezes amolecidas. São também relativamente normais alterações no metabolismo dos lipídeos, com o aumento do colesterol e triglicérides, e da glicose, com intolerância a glicose ou, mais raramente, até mesmo desenvolvimento de diabetes (LOPES, 2007).
As interações medicamentosas potenciais são extensas. A co-administração com rifampicina, carbamazepina, fenobarbital, fenitoína, dexametasona ou erva-de-são joão pode reduzir os níveis de LPV/r (KATZUNG, 2006).
Cada comprimido é formulado com 133,3 mg de LPV/r e 33,3 mg de RTV. A dose recomendada para adultos é de 400 mg de LPV/r, associada a 100 mg de ritonavir a cada doze horas. O uso concomitante de EFZ ou NVP requer a elevação da dose para quatro comprimidos a cada dose horas, por causa do aumento na velocidade de metabolização do LPV/r pelo fígado. Também disponíveis em forma de suspensão para uso por criança ou mesmo adultos (LOPES, 2007).
Amprenavir
O amprenavir é inibidor de protease viral com especificidade para o vírus HIV, cujo perfil de resistência a esse fármaco parece ser diferente do de outros congêneres, pois sua eficácia permanece é por mais de 24 horas. Esta é a razão para dose única diária (MOTA et al., 2004). É rapidamente absorvido pelo trato gastrointestinal, podendo ser ingerido com ou sem alimentação. Todavia, as refeições ricas em gordura podem diminuir a absorção e, portanto, devem ser evitadas. A ligação às proteínas é de cerca de 90% (KATZUNG, 2006). É altamente dependente do metabolismo pela enzima CYP3A4, do complexo enzimático CYP450. Com meia-vida de nove horas, é empregado na dose de 1.200 mg a cada dose horas ou e associação com RTV, que eleva o pico e a meia-vida sérica do amprenavir (1.200 mg de amprenavir e 200 mg de RTV em dose única diária ou divididos em duas tomadas ao dia) (LOPES, 2007).
A base da resistência in vitro ao amprenavir parece ser a mutação I50V. As evidências disponíveis até o momento sugerem que a resistência cruzada a outros membros da classe de IPs pode ser menos prevalente com o amprenavir do que com os componentes previamente disponíveis (KATZUNG, 2006).
Estudos clínicos mostram como principais efeitos colaterais do amprenavir: erupção cutânea, cefaléia, diarréia, exantema e perda de peso, mas pouco se sabe sobre as repercussões desse fármaco sobre a gestação (MOTA et al., 2004). O exantema é relativamente freqüente (cerca de 22% dos pacientes), aparecendo ao redor do décimo primeiro dia de uso. Costuma ser benigno e pode desaparecer mesmo com a manutenção da droga, mas leva a interrupção de seu uso em 3% dos pacientes, e 1% dos pacientes pode apresentar casos mais graves, incluindo síndrome de Stevens-Johnson (LOPES, 2007). O amprenavir é tanto substrato quanto inibidor da CYP3A4, e sua administração esta contra indicada com numerosos outros fármacos. A solução oral, que contem propilenoglicol, está contra-indicada para crianças de pouca idade, mulheres grávidas e pacientes em uso de metronidazol ou dissulfiram (KATZUNG, 2007). O amprenavir está disponível também, em comprimidos de 150 mg. O esquema de duas tomadas de 600 mg associados a 100 mg de ritonavir tem merecido preferência relativamente ao seu uso isolado (LOPES, 2007).
Atazanavir - ATV
Aprovado para uso em 2004, sulfato de atazanavir pode ser utilizado como único IP do esquema ARV em pacientes no início do tratamento ou em associação com RTV, quando empregado em esquemas de resgates. Com boa absorção oral, melhorada quando administrado com alimentos, possui longa meia-vida, o que permite seu uso em dose única diária. O ATV não deve ser administrado com inibidores de bomba de prótons, como o omeprazol e pantoprazol, pois estes diminuem sua concentração e reduzem a eficácia terapêutica. Deve ser administrado com cautela em associação a inibidores de receptores H2, quando é recomendado intervalo de doze horas entre ambos, e antiácidos, quando é recomendado intervalo de duas horas antes ou uma hora após o uso dessa drogas (LOPES, 2007).
È também metabolizado pela enzima CYP3A4. É primariamente eliminado pelas fezes, e pode ser usado em pacientes com insuficiência renal. Os efeitos colaterais associados ao uso de ATV são prolongamento do intervalo PR (um indicativo da velocidade de condução entre os átrios e os ventrículos), que demanda cuidado em pacientes com distúrbios de condução atrioventricular ou uso de outras drogas que aumentam o intervalo PR (como beta bloqueadores, diltiazem, verapamil e digoxina), desencadeamento ou exacerbação de intolerância a glicose e diabetes, aumento de episódios de sangramento em hemofílicos, exantema e sintomas gastrintestinais em elevação. O ATV é também um inibidor da enzima uridina difosfato glucuronil transferase, levando na deficiência na conjugação da bilirrubina e icterícia em significativa proporção de pacientes, especialmente aqueles com síndrome de Gilbert, mas não traz riscos ao paciente e é reversível após a suspensão da medicação (LOPES, 2007).
Formulado em comprimidos de 150 a 200 mg, deve ser administrado uma vez ao dia, na dose de 400 mg (quando único IP) ou 300 mg (quando associado com 100 mg de RTV). O ATV não deve ser utilizado como único IP em esquemas que contenham tenofovir ou EFZ, quando deve ser empregado na dose de 300 mg em associação com 100 mg de RTV (LOPES, 2007).
Fosamprenavir - FPV
É uma pró-droga, facilmente hidrolisada em seu componente ativo, o amprenavir, com vantagens farmacocinéticas que permitem melhor posologia e menor carga de comprimidos. Como ocorre com outros IPs do HIV-1, o FPV liga-se ao sitio ativo da protease do HIV-1, impedindo o processamento dos precursores virais das poliproteínas Gag e Gag-Pol e resultando na formação de virions imaturos não infecciosos. O FPV não possui algumas limitações que estão associados ao amprenavir, incluindo uma cápsula grande, uma grande quantidade de comprimidos a ingerir e a formulação em cápsula de gelatina. Os comprimidos podem ser administrados independentemente do horário das refeições. A ligação do amprenavir a proteínas plasmáticas é de aproximadamente 90%, ligando-se primariamente à glicoproteína α1-ácida (TORRES & ARDUINO, 2007).
A principal via de eliminação do amprenavir é o metabolismo hepático através do sistema enzimático CYP450, primariamente por oxidação e em menor grau, por conjugação. A maioria das interações medicamentosas clinicamente relevantes é devida à inibição da CYP450 pelo amprenavir (TORRES & ARDUINO, 2007).
Três dosagens de FPV foram aprovados pelo FDA para o tratamento de pacientes com HIV-1 sem tratamento prévio: FPV 1.400 mg duas vezes ao dia, FPV 1.400 uma vez ao dia + ritonavir 200 mg uma vez ao dia e FPV 700 mg duas vezes ao dia + ritonavir 100 mg duas vezes ao dia. Nos pacientes infectados pelo HIV-1 previamente tratados com ARV, somente uma dosagem foi aprovada: FPV 700 mg duas vezes ao dia + ritonavir 100 mg duas vezes ao dia. A droga é fornecida em comprimidos de 700 mg (TORRES & ARDUINO, 2007).
Os efeitos colaterais e interações medicamentosas são comparáveis ao amprenavir (LOPES, 2007). As principais mutações da protease do HIV-1 que conferem resistência ao FPV incluem a I50V e a I84V. as mutações menores incluem a L10F/I/R/V, a V32I, a M46I/L, a I47V, a I54L/V/M, a G73S e a L90M (TORRES & ARDUINO, 2007).
Tipranavir
Aprovado para uso nos EUA em 2005 constituiu uma novo IP não peptídico com potencial para uso em pessoas infectadas por vírus resistentes a outros representantes da classe de IPs. Deve ser administrada, juntamente com baixas doses de RTV para obtenção de níveis sanguíneos adequados. O tipranavir também é metabolizado pela enzima CYP3A4, sendo, também, um indutor desta enzima e da glicoproteína P, ambas inibidas pelo RTV. A interação de ambos, portanto, é relativamente complexo, o que torna o uso de tipranavir com os demais IPs não recomendável até o momento. É formulado em cápsula de 250 mg e a dose aconselhável de tipranavir é de 500 mg duas vezes ao dia, em associação a 200 mg de RTV. Não requer ajuste em pacientes com insuficiência renal (LOPES, 2007).
Como os outros IPs, o tipranavir pode induzir sintomas gastrintestinais, como diarréia, náuseas, vômitos e dor abdominal. Induz aumento de transaminases com maior freqüência que outros IPs, requerendo monetarização cuidadosa da função renal em pacientes co-infectados pelos vírus das hepatites B e C e com outras doenças hepáticas. Por conter um resíduo de sulfonamida, o tipranavir pode levar a exantema alérgico e necessita ser usado com cuidado ou evitado em pacientes com historia de reação a sulfa (LOPES, 2007).
Darunavir
O darunavir é projetado para ser ativa contra o HIV resistente a IPs disponíveis atualmente. Durante seu desenvolvimento, era conhecida como TMC114. O darunavir foi concedido à aprovação acelerada para uso nos Estados Unidos em junho de 2006.
Disponível para o tratamento de pacientes nos Estados Unidos, cuja infecção por HIV não está respondendo ao tratamento com outros medicamentos anti-HIV (AIDS MAP, 200?).
Não existem dados disponíveis sobre a segurança e a eficácia do darunavir em doentes que não tenham tomado quaisquer outros medicamentos anti-HIV antes. O efeito do dano hepático sobre a eficácia do darunavir é ainda desconhecida (AIDS MAP, 200?).
A dose padrão de darunavir é 600mg duas vezes ao dia, tomado com RTV 100mg para aumentar os níveis de darunavir no sangue. O darunavir deve ser tomado juntamente com alimentos, para garantir níveis adequados da droga no sangue. O darunavir é fabricado em comprimidos laranjas, sendo que cada comprimido contém 300mg de drogas (AIDS MAP, 200?).
Os efeitos colaterais mais comuns são diarréia, náuseas e dores de cabeça, mas são menos freqüentes que outros IPs. Cerca de 7% dos pacientes pode apresentar erupção cutânea, grave, em alguns casos (AIDS MAP, 2008).
A grande mutação de resistência ao darunavir observadas em estudos é V32I. Outras mutações como I54L/M e I47V também são comuns. Os pesquisadores acreditam que um certo número de mutações são necessárias de se acumular antes de uma resposta reduzida ao medicamento. Há pouca resistência cruzada entre darunavir e outros IPs (AIDS MAP, 2008).
O darunavir é metabolizado CYP3A4 no fígado. Portanto existem interações significativas com outros fármacos que são metabolizados por esse sistema, como Carbamazepina, Cisaprida, Midazolam, Fenobarbital, Rifamicina, Triazolam. Mulheres usando contraceptivos hormonais como estradiol podem apresentar níveis reduzidos. Métodos alternativos de contracepção são recomendados (AIDS MAP, 2008).
4.2.4 Inibidores da Enzima Integrase
A enzima integrase é fundamental no processo de replicação viral, sendo responsável pela integração do DNA viral ao cromossomo hospedeiro, permitindo assim a continuação do ciclo da replicação viral (SOUZA & ALMEIDA, 2003).
Aparentemente estes compostos têm a capacidade de prevenir o processo de integração mesmo após a formação do chamado complexo pré-integrativo, formado pelo DNA viral, integrase e outras proteínas (PEÇANHA et al., 2002).
Raltegravir
Raltegravir, também conhecido como MK-0518, é um dos primeiros de uma nova classe de ARV, os inibidores da integrase. A inibição da integrase impede a inserção do DNA do HIV no DNA do genoma humano, assim bloqueando a capacidade do HIV em se replicar. Raltegravir foi aprovado pela FDA em 2007, para ser utilizado com outros agentes ARV no tratamento da infecção pelo HIV. Este medicamento recebeu aprovação para o uso em doentes adultos experientes em tratamentos ARV que tenham provas de replicação viral de HIV-1 e estirpes resistentes a múltiplos regimes ARV. A análise de ensaios clínicos com raltegravir, conduziu a reduções significativas da carga viral do HIV e aumenta da contagens de células CD4 (AIDS INFO, 2008).
O modo de ingerir é por via oral. A forma de apresentação é em comprimidos contendo 400 mg de raltegravir. A dose recomendada de raltegravir para adultos experientes no tratamento ARV infectados pelo HIV é um 400 mg duas vezes ao dia. Não é necessário um ajuste posológico em doentes com insuficiência hepática ou insuficiência renal grave. Uma refeição rica em gorduras modificou o ritmo de absorção, resultando em uma diminuição de aproximadamente 34% na concentração plasmática máxima (Cmax), um aumento de 8,5 sobre a concentração plasmática às 12 horas, e um atraso no tempo até à concentração máxima (Tmax) após uma dose única 400 mg (AIDS INFO, 2008).
Nenhuma evidência de mutagenicidade ou genotoxicidade foi observada durante os testes de mutagênese microbiana in vitro. Não tem efeito sobre a fertilidade, observado em estudo com ratos machos e fêmeas em doses até 600 mg/kg/dia, o que resultou em uma exposição superior a três vezes a exposição humana em relação a dose recomendada. As concentrações plasmáticas de raltegravir no feto são de aproximadamente 2% da média da concentração materna, tanto de 1 a 24 horas após a dose de 1000mg / kg / dia, comprovadas em estudo com coelhas. Raltegravir deve ser utilizado durante a gravidez somente se claramente necessário. Não se sabe se o raltegravir ou de seus metabólitos são distribuídos para o leite humano (AIDS INFO, 2008).
Cerca de 83% do raltegravir se liga a proteínas plasmáticas durante o intervalo de concentração de 2 a 10 MCM. A meia-vida terminal aparente de raltegravir é de aproximadamente 9 horas, com um curta fase de semi-vida (cerca de 1 hora). Após a administração de uma dose oral de raltegravir, aproximadamente, 51% e 32% da dose é excretada nas fezes e urina, respectivamente. Dois componentes, raltegravir e raltegravir - glucuronido, são detectadas na urina e representaram cerca de 9% e 23% da dose, respectivamente. Dois Dados indicam que o principal mecanismo de depuração do raltegravir nos seres humanos é mediada pela UGT1A1 (Uridina difosfato glucuronosiltransferases) (AIDS INFO, 2008).
Pelo menos 50% dos pacientes tratados com raltegravir, podem reduzir níveis de carga viral de 400 cópias/ml para, pelo menos 50 cópias/mL. Em um estudo comparando doses de efavirenz e raltegravir, demonstrou que pacientes recebendo tratamento com raltegravir em qualquer dose, apresentaram cargas virais mais rapidamente indetectáveis, comparadas ao tratamento com efavirenz (AIDS INFO, 2008).
A falha virológica pode ocorrer em 3% dos pacientes e pode estar associado ao aminoácido N155H. Raltegravir nas concentrações de 6 a 50 nM resultou na inibição de 95% (EC95) da disseminação vírica do sangue periférico humano infectado com diversos isolados clínicos primários do HIV-1, incluindo isolados resistentes aos ITRs e IPs. Alterações da sinérgica da atividade ARV foi observada quando células T humanas infectadas com o H9IIIB, variante do HIV-1, foram incubadas com raltegravir em combinação com os ITRNNs (delavirdina, efavirenz e nevirapina),ITRNs (abacavir, didanosina, estavudina, tenofovir, zalcitabina, e zidovudina), IP (amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir e saquinavir), ou o IsF enfuvirtide (AIDS INFO, 2008).
Os efeitos adversos provocados pelo raltegravir são diarréia, náuseas, fadiga, dores de cabeça, e prurido. Outros efeitos adversos relatados incluíram obstipação, flatulência, e sudorese. Raltegravir deve ser utilizado com precaução quando administrado com indutores de UGT1A1, incluindo rifampicina. Estes indutores do UGT1A1 pode reduzir as concentrações plasmáticas de raltegravir (AIDS INFO, 2008).
4.2.5 Inibidores de Fusão (IsF)
Os inibidores de fusão (IsF) representam uma nova abordagem na estratégia de combate à capacidade de replicação do HIV no organismo (SOUZA & ALMEIDA, 2003), e são utilizadas como terapia de resgate (LAVRA, 2006). Para que o HIV complete o seu ciclo reprodutivo, necessita se fundir com um linfócito T, onde deposita a sua informação genética, dando origem a novos vírus. Os IsF foram concebidos de forma a impedir que o vírus consiga penetrar nos linfócitos e nem sequer inicie a infecção. Os compostos concebidos são capazes de bloquear três tipos de interação: 1) bloquear a interação da gp 120 com o CD4; 2) bloquear a interação da gp 120 com os co-receptores; 3) inibir as interações com a gp 41 (SOUZA & ALMEIDA, 2003).
4.2.5.1 Bloqueadores da Interação da gp 120 com o CD4
Pro 542
PRO 542 é uma proteína que foi concebida para inibir a entrada de HIV através da interação com a região gp120 do HIV com receptores CD4 na superfície dos linfócitos. É produzido a partir do vírus vinculando domínios da molécula CD4, fundidas a imunoglobulina humana G. Também é conhecido como recombinante CD4-IgG2. PRO 542 é o último na linha de receptores CD4 solúvel, um grupo de agentes que mostraram pouca eficácia quando testada nos anos antes do surgimento da TARV. Está atualmente atravessando fase II nos ensaios clínicos nos Estados Unidos. Após uma única dose, o estudo concluiu que o composto foi bem tolerado e resultou em reduções da carga viral de até 2 log10. Redução da carga viral foi dependente da dose.
Também foi apontado que o vírus infeccioso não pode ser recuperado por mais de 72 horas após uma única perfusão, levando os investigadores a sugerir que o PRO 542 pode ter valor especial como uma profilaxia contra a infecção pelo HIV, quer em tratamentos perinatais ou após exposição sexual (AIDS MAP, 2008).
PRO 542 é sinérgica com a T-20 in vitro; doses de cada uma das drogas podem ser reduzidas em dez vezes, se forem utilizados em conjunto, sendo que a replicação viral foi reprimida tão bem juntas do que se uma ou outra droga tivesse sido utilizada isoladamente. Os pesquisadores observam que esta sinergia pode permitir que o esquema posológico de cada medicamento seja simplificado, cuja principal preocupação é com os produtos que têm de ser administrados por injeção ou perfusão (AIDS MAP, 2008).
Uma neutropenia de grau três foi relatado em um paciente quatro semanas após uma única perfusão de PRO 542, e os únicos eventos adversos relatados em estudos pediátricos foi uma febre em uma criança com histórico prévio de tais reações a infusões de imunoglobulina. PRO 542 deve ser injetado, pois é uma proteína que seria desnaturada no estômago. Uma dose escalada de perfusão intravenosa é utilizada para definir a dose mais elevada tolerada, e estudos pediátricos têm utilizado perfusões intravenosas quatro vezes por semana para efetivar a tolerabilidade da droga. A meia-vida da droga após uma única infusão é de 3,3 a 4,2 dias, sugerindo que pode não ser necessária À infusão deste composto todos os dias (AIDS MAP, 2008).
A dosagem de uma ou duas vezes por semana pode ser possível em doses superiores a 10mg/kg. Na fase I / II com estudos em crianças a dose varia, PRO 542 foi administrada uma vez por semana, e os níveis in vitro de IC 50 foram mantidos elevados por aproximadamente cinco dias. 542 PRO não parece ser imunologicamente reativo, anticorpos contra a proteína não foram detectados (AIDS MAP, 2008).
BMS 488043
BMS 488043 ou 043 é um fármaco experimental anti-HIV em desenvolvimento que visa prevenir a ligação da molécula gp120 do HIV ao receptor CD4. Ao bloquear gp120, BMS 488043 pode ser capaz de prevenir que células T CD4 fiquem infectadas com o HIV. Como um inibidor da entrada que visa à interação entre o vírus e o receptor CD4 primário, 043 é eficaz contra o vírus que quer utilizar os co-receptores CCR5 ou CXCR4. Estudos preliminares da BMS 488043, foram conduzidos em pessoas HIV- positivos e HIV- negativo. É administrado em doses de 1200 e 1800mg duas vezes ao dia com uma refeição rica em gorduras (AIDS MAP, 2008).
O pico de redução da carga viral é observada no nono dia de tratamento, com média de 1,23 log10 na dose de 1800mg e 1,01 log10 na dose de 800mg, embora a carga viral se recupere lentamente na maioria dos pacientes. A média da contagem de células CD4 fica por volta de 106 células/mm3. Eventos adversos e anomalias laboratoriais são leves (AIDS MAP, 2008).
Pesquisas tem fornecido algumas informações preliminares sobre o perfil de resistência provável da BMS 488043. Mutações resistente de BMS 488043 ocorrem principalmente dentro do envelope na proteína gp120. Eles incluem V68A, M426L, M434I, S440R, e M475I. Mutações M426L ou M475I, que são próximos do ponto de contato ao receptor CD4, conferem os mais altos níveis de resistência. Outra mutação é no sitio de contato, através da W427V, seletivo para o receptor CD4 a partir de gp120, que também elimina a ligação do fármaco a gp120. A mutação S375W, aprofunda a cavidade vinculativa, obrigando a diminuição da BMS 488043. Isso demonstra que a gp120 se liga à droga em uma região específica, que sobrepõe com o sitio vinculativo gp120-CD4 (AIDS MAP, 2008).
4.2.5.2 Bloqueadores da interação da gp 120 com os co-receptores
Maraviroc
Maraviroc é um antagonista dos receptores de quimiocina. Foi concebido para prevenir a infecção pelo HIV de células T CD4 por bloquear o receptor CCR5. Quando o receptor CCR5 está indisponível, o HIV não pode penetrar a membrana para infectar as células CD4. Durante seu desenvolvimento, era chamado RU- 427, 857. É o primeiro agente em sua classe a ser testada em pessoas com infecção pelo HIV. Os resultados preliminares de um estudo de Fase I apresentados em 2003 onde se administrou nos pacientes 25mg, uma vez ao dia ou 100mg duas vezes ao dia de maraviroc como monoterapia, demonstrou que o estado estacionário dos níveis da droga foi atingido dentro de sete dias, com níveis mais elevados da droga em pacientes que tiveram jejum. Demonstrou ainda que no décimo quarto dia a dose de 100mg sofre um declínio na carga viral de 1,4 log10 e 0,4 log10 na dose de 25mg (AIDS MAP, 2008).
O CCR5 é um alvo especialmente interessante, pois a ausência do mesmo, resulta em quase completa, a resistência à infecção pelo HIV-1 (MACARTHUR & NOVAK, 2008). Maraviroc foi encontrado para ter potente atividade antiviral in vitro contra todas as linhagens virais CCR5-trópicas testado, incluindo 43 isolados a partir de várias clades primárias e diversificada origem geográfica (média MIC90, 2,0 nmol/L) (MACARTHUR & NOVAK, 2008).
O composto tem um peso molecular de 514 g/mol e é uma molécula moderadamente lipofílica (log D7.4 valor, 2.1) e básica (pKa, 7.3). A Absorção de maraviroc é rápida, mas variável, com o tempo de absorção máxima sendo geralmente 1 - 4 horas após a ingestão do fármaco.A farmacocinética do maraviroc oral não é proporcional à dose acima do intervalo posológico. A biodisponibilidade absoluta é de 23% para uma dose de 100 mg e se prevê que venha a ser 33% de uma dose de 300 mg. A meia-vida de eliminação após uma única dose oral de 300 mg é 10.6 + 2.7 h. A meia-vida terminal de maraviroc após a administração de uma dose oral consecutiva de um estado estacionário em indivíduos saudáveis foi 14 - 18 h (MACARTHUR & NOVAK, 2008)
A droga é bem tolerada, e a carga viral não sobe de imediato após a cessação da droga, o que indica que uma proporção de receptores permanecem bloqueados por algum tempo. A dose de 300mg duas vezes ao dia parece ser a opção mais promissora para o tratamento futuro, apesar que 300 e 600mg, uma vez por dia estão também a ser investigada em ensaios atuais (AIDS MAP, 2008).
Uma vez que maraviroc liga-se à co-receptor CCR5, tem havido alguma preocupação de que o seu uso em pacientes com HIV trópicos de R5 podem estimular uma conversão para o HIV que usa como alternativa o co-receptor CXCR4 (AIDS MAP, 2008). Maraviroc é um substrato da enzima CYP3A4, e tem potenciais interações com outros medicamentos que são metabolizados por esta enzima, especialmente IPs. Por exemplo, os níveis de maraviroc estão aumentadas em pacientes tomando também ATZ, LPV/r e RTV/SQV (AIDS MAP, 2008).
4.2.5.3 Inibidores da interação com a gp 41
Enfuvirtida (T-20)
A enfuvirtida representa a primeira droga de uma nova classe de ARV, os IsF. Age de forma competitiva, inibindo a interação entre os domínios HR1 e HR2, necessários para a fusão viral (TENORE & FERREIRA, 2007). A T-20 mimetiza a estrutura da gp41, responsável pela fusão e adesão do vírus HIV à célula CD4. Como a ação do fármaco é extracelular, pode-se obter o sucesso terapêutico mesmo em pacientes com vírus resistentes a todos os outros ARV. A administração de T-20 não é oral porque se trata de um peptídeo, sendo rapidamente digerido no trato gastrintestinal. Estudos mostraram que a via para administração subcutânea é mais efetiva que a intramuscular, e, é bem tolerada, mantendo as concentrações de T-20 em estado de equilíbrio durante 12 horas após a injeção SC. Em estudo fase I e II, a monoterapia com T-20 resultou em uma rápida redução de carga viral, média de 1,96 log10 cópias/ ml depois de 14 dias de tratamento (SANTOS, 2006). A ligação às proteínas é elevada (92%) e o metabolismo parece ocorrer através de hidrolise proteolítica, sem a participação do sistema CYP450 (KATZUNG, 2006).
Pode ser administrado a pacientes com insuficiência renal leve e moderada (clearance de creatinina maior que 35 mL/minuto), mas não há dados para uso em pacientes com insuficiência renal grave ou hepática (LOPES, 2007).
O principal evento adverso é a formação de nódulos subcutâneos, dolorosos no local da aplicação (TENORE & FERREIRA, 2007), alem do desconforto, eritema, prurido e equimose, com especial atenção às infecções locais (LOPES, 2007). Os eventos adversos sistêmicos que ocorrem com mais freqüência são diarréia, náuseas e fadiga (SANTOS, 2006). Os estudos com T-20 demonstram também aumento da incidência de pneumonia. Não há uma explicação clara para essa associação, mas obriga a monetarização de sinais e sintomas de pneumonia (LOPES, 2007). Raramente, pode causar reação complexa primaria do sistema imunológico, desconforto respiratório, glomerulonefrite e síndrome de Guillain-Barre (LOPES, 2007).
Pode ocorrer resistência a T-20, e tanto a freqüência quanto os mecanismos desse fenômeno estão sendo atualmente investigados. Todavia, a T-20 carece por completo de resistência cruzada com outras classes de fármacos ARV atualmente aprovados (KATZUNG, 2006).
Atualmente, esta droga serve como opção de resgate para pacientes com múltiplas falhas e que tenham, pelo menos, uma ou duas drogas ativas indicadas pelo exame de genotipagem (SANTOS, 2006). A dose recomendada é de injeções subcutâneas contendo 90 mg de T-20 a cada 12 horas (LOPES, 2007).
T - 1249
Com o objetivo de se obter IsF mais potentes a diferentes tipos de HIV resistentes, foi concebido o T-1249 26, um peptídeo sintético de 39 aminoácidos, que representa uma geração mais recente do T-20. Estudos preliminares indicam que esta droga é cerca de 100 vezes mais ativa do que o T-20, sendo sua posologia indicada como dose única diária, injetável. Atualmente, o T-1249 se encontra em fase de testes clínicos I/II (SOUZA, 2005).
4.3 ESQUEMAS TERAPÊUTICOS PARA O INICIO DO TRATAMENTO
Com maior numero de medicamentos hoje disponíveis, a escolha do esquema inicial tornou-se um problema mais complexo, em particular para o não especialista (LOPES, 2007).
A terapia inicial deve sempre incluir combinações de três drogas: dois ITRN associados a um ITRNN ou a um IP reforçado com RTV (IP/r). Os IPs potencializados com RTV (IP/r) oferecem maior barreira genética à resistência que os ITRNN. Isto significa que para que se desenvolva resistência a um IP/r, há necessidade de um numero maior de mutações que para o desenvolvimento de resistência a TRNN. De fato, a resistência a qualquer IP/r resulta do acumulo de mutações, enquanto apenas uma mutação de ITRNN confere resistência completa ao EFV e NVP. Em conseqüência a barreira genética dos fármacos, a barreira genética dos esquemas contendo IP/r também é maior. Realmente, a comparação de LP/r com EFV em esquema de terapia inicial mostra que a falha virológica (decorrente de mutações de resistência) é mais freqüente com o esquema baseado em EFV (BRASIL, 2007).
Achados de metanálise de 53 ensaios clínicos randomizados avaliando a terapia inicial em 48 semanas de seguimento mostram equivalência na proporção da resposta virológica ao esquema inicial entre pacientes que recebem 2 ITRN + ITRNN (EFV) e 2 ITRN + IP/r. Um ensaio clinico randomizado que comparou diretamente esquemas iniciais de TARV, combinações envolvendo 2 ITRN e ITRNN (EFV) com 2 ITRN e IP/r (LPV/r) demonstrou que, na analise de intenção de tratamento, os resultados de supressão viral (carga viral < 50 cópias/mL) nos pacientes do grupo EFV (89% de supressão viral) foram superiores ao grupo LPV/r (77%) (BRASIL, 2007).
Particularmente, em estratégias de terapia seqüencial, não existem dados publicados de longo prazo que permitam definir qual é a abordagem associadas com melhores resultados. Por outro lado, vários estudos comparativos envolvendo pacientes virgens de tratamento, mostram que as taxas de sucesso virológico (medido pela proporção de indetectabilidade viral) nos esquemas contendo 2 ITRN + ITRNN foram, na maioria, equivalentes às taxas obtidas nos grupos que usaram esquemas contendo IP ou IP/r (BRASIL, 2007)
Alem disso, esquemas que utilizam 2 ITRN + ITRNN são, em geral, de posologia mais simples o que provavelmente facilita a adesão ao tratamento, Adicionalmente, a longa meia-vida do EFV permite flexibilidade no horário de tomada (BRASIL, 2007).
O paciente deve ter clareza sobre a importância do primeiro esquema ARV, como o momento com maior possibilidade de supressão da replicação viral e da resposta imunológica. O quadro 1 define os critérios para inicio da terapia em pacientes infectados pelo HIV (BRASIL, 2007).
.jpg)
Fonte: (BRASIL, 2007)
A presença de sintomas ou manifestações clinicas associadas a imunodeficiência relacionada ao HIV, mesmo quando não definidoras de AIDS, sugere a necessidade de iniciar o tratamento ARV, independente dos parâmetros imunológicos, devendo esta decisão ser considerada individualmente (BRASI, 2007).
4.4 FALHA TERAPÊUTICA
Define-se falha terapêutica quando, na vigência de tratamento ARV, o paciente apresentar qualquer infecção oportunista (falha clinica); quando houver queda progressiva na contagem de celular CD4+ (falha imunológica); quando houver aumento repetido da carga viral (geralmente aumento maior que 0,5 log ou contagem superior a três vezes ou mais) ou quando a mesma permanecer constantemente elevada, ou seja, acima de 400 cópias/mL (falha virológica) (FOCACCIA & VERONESI, 2007).
Quando um paciente que inicia o tratamento é avaliado, devemos considerar vários fatores, como história clinica, exame físico, resultados da carga viral e contagem de linfócitos T CD4+ (preferencialmente em dois momentos após o inicio do tratamento), capacidade do paciente seguir as orientações médicas, aderência ao tratamento, opções remanescentes, potencial presença de cepas virais antes do inicio do tratamento e participação do paciente nas decisões que resultam em manutenção ou modificação do tratamento (LOPES, 2007). A falta de adesão do paciente ao tratamento, absorção inadequada do medicamento, interação entre as drogas utilizadas, pouca potencia antiviral do medicamento e conseqüente adquirição de resistência viral as drogas, podem acarretar em falha terapêutica (FOCACCIA & VERONESI, 2007).
Estudos demonstram que um significativo número de pacientes apresenta falência as combinações de drogas, em virtude do desenvolvimento da resistência genotípica do HIV-1. Em um estudo em São Paulo, foram avaliados 791 pacientes, os quais, ao serem submetidos a exames de genotipagem do HIV, apresentaram falha a terapia HAART. Os resultados evidenciaram que 96,6% apresentaram resistência relacionada ao gene da enzima TR, 90,3% a Protease e 99,4% a ambas (KALMAR, 2007). A falha terapêutica esta bastante relacionada com resistência primaria aos ARV. Em um estudo clinico retrospectivo com pacientes tratados com FTC, ddI e EFV comparados com d4T, ddI e EFV encontrou 16% de resistência primaria aos ITRN ou TRNN e a falha foi significativamente maior no grupo de pessoas com resistência primaria (CASEIRO & DIAZ, 2007).
Ao se optar pela troca de tratamento, cabe ao médico e ao paciente a preferência por uma combinação de drogas para resgate. A escolha dos medicamentos deve ser realizada baseada na circunstancia em que ocorreu a falha do tratamento inicial. Em casos de falha por dificuldade de ação dos ARV por pobre aderência, ma absorção ou interação medicamentosa, deve-se corrigir a causa base ou trocar o tratamento pra facilitar a aceitação e aderência do paciente (LOPES, 2007).
O aumento na carga viral na vigência de tratamento é a melhor expressão de emergência de cepa viral resistente. A resistência viral é demonstrada pela presença de mutações nos genes que expressam a TR ou a protease. A caracterização destas mutações é feita pelo teste de genotipagem do HIV para os genes acima (FOCACCIA & VERONESI, 2007).
4.5 RESISTÊNCIA AOS ANTI-RETROVIRAIS
A resistência viral é definida pela presença de mutações no genoma viral que reduzem a suscetibilidade à droga quando comparada ao vírus selvagem. As mutações que conferem resistência aos anti-retrovirais são selecionadas pela terapia (MANENTI, 2008).
As mutações podem ser classificadas como: principais - quando sozinhas conferem resistência, acessórias - que podem alterar a capacidade replicativa do vírus na presença de mutações principais, primárias - quando surgem em pacientes virgem de tratamento e secundárias – que aparecem nos pacientes em uso de antiretrovirais (MANENTI, 2008).
Em relação aos ITRN têm-se dois mecanismos de resistência descritos até o momento: mutações que promovem a capacidade ao vírus de distinguir entre dNTP fisiológico e o ITRN, como por exemplo, M184V, Q151M e K65R e mutações que promovem a excisão hidrolítica do ITRN incorporado a fita de cDNA , são denominadas TAMs e ocorrem nos códons 41, 67, 70, 210, 215 e 219 da transcriptase reversa. As mutações que conferem resistência aos ITRNNs reduzem a afinidade pelo medicamento, por exemplo, G190A. As mutações de resistência aos IP modificam a quantidade e natureza dos pontos de ligação entre os inibidores e a enzima (MANENTI, 2008).
Testes como fenotipagem, genotipagem e fenótipagem virtual podem identificar cepas resistentes. O seqüenciamento genético do gene pol (PT e RT), a partir do PCR direto permite detectar mutações associadas a resistência genética na população viral majoritária (MANENTI, 2008).
4.6 TESTE DE GENOTIPAGEM
Existem duas classes de testes laboratoriais para detectar resistência aos ARVs; os testes para determinação de resistência fenotípica ou fenotipagem e os testes para determinação de resistência genotípica ou genotipagem (DIAZ, 2006). Ambas são ferramentas muito utilizada após falha na terapia ARV, elas mostram as principais mutações aos ITRNs, ITRNNs e IP. Sabe-se que o custo e o tempo de execução dos métodos são compensados com a substituição direcionada do fármaco, reduzindo os custos, os efeitos adversos e principalmente o aparecimento de mais mutações com resistência as TARVs (CORREIA, 2005).
Os testes genotípicos determinam a seqüência genômica da região que codifica a transcriptase reversa e a protease do HIV-1. Dentre as abordagens existentes temos a reação em cadeia pela polimerase (PCR) seletiva, PCR com hibridização pelo uso de sondas (LIPA) e seqüenciamento genômico. Os métodos que utilizam o seqüenciamento promovem uma avaliação mais ampla e especifica. Dentre as metodologias temos o seqüenciamento manual clássico, o seqüenciamento automatizado (ABI, Pharmacia) e o seqüenciamento em microchip por hibridização (affymetrix). Existem atualmente kits comerciais padronizados e licenciados pelo FDA, como os kits comercializados pela Bayer e pela Abbott (DIAZ, 2006).
A tradução em aminoácidos se dá de forma que cada trinca de nucleotídeos codificará um aminoácido, e a interpretação disto vai gerar um laudo típico de genotipagem (DIAZ, 2006).
4.7 TERAPIAS DE RESGATE
Atualmente, devido a diversos fatores, incluindo intolerância e/ ou má adesão ao tratamento, uso prévio de esquemas inadequados e, mais raramente, resistência primaria, há uma parcela de pacientes que apresenta vírus resistentes e que necessita de novos esquemas ARV, denominados “terapia de resgate” (BRASIL, 2007).
Distintamente do que ocorre em relação à terapia inicial, há escassez de recomendações consensuais especificas para escolha de esquemas de resgate. Isso se deve à relativa carência de ensaios clínicos randomizados (ECR) que tenham comparado diferentes estratégias de resgate recrutando grande número de pacientes. Nos últimos dois anos, no entanto, resultados de vários ensaios clínicos abordando o manejo de pacientes com múltiplas falhas prévias foram publicados. Embora tenham contribuído para o conhecimento sobre a terapia de resgate, todos foram desenhados para abordar a eficácia de novas drogas (inibidores de protease potentes ou drogas de novas classes) em pacientes com ampla experiência prévia com anti-retrovirais, portadores de vírus multirresistentes (BRASIL, 2007).
Sendo assim, as recomendações para terapia de resgate baseiam-se por vezes em inferências teóricas, estudos pilotos ou subanálises de estudos clínicos desenhados para outras finalidades. Feitas essas ressalvas, reconhece-se que algumas recomendações são essenciais para orientar o clínico e podem auxiliá-lo na implementação de um esquema anti-retroviral com maior chance de eficácia para o paciente já tratado previamente (BRASIL, 2007).
No Quadro 2, Seguem algumas dicas importantes a serem consideradas na avaliação da resistência genotípica que devem orientar a escolha dos esquemas de resgate. Não são regras absolutas, já que o grupo de pacientes em falha terapêutica é bastante heterogêneo em relação a causas de falha, número e tipo de esquemas anti-retrovirais prévios, prevalência de mutações de resistência, opções de drogas ativas, limites e possibilidades do novo esquema etc. Assim, em situações em que há dúvida quanto a esses princípios e ao manejo do caso, recomenda-se que o médico assistente lance mão da retaguarda técnica existente, levando o caso à discussão com os Médicos de Referência em Genotipagem (MRG), câmaras técnicas ou serviços de referências que detenham ampla experiência no tratamento de pacientes com múltiplas falhas de tratamento prévias e portadores de vírus multirresistentes (BRASIL, 2007).
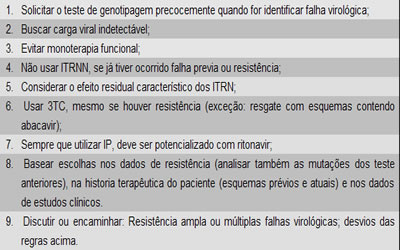
Fonte: (BRASIL, 2007)
A melhor forma de orientar um novo tratamento, especialmente na primeira falha, é baseá-lo no teste de genotipagem. Nas regiões do país com menor acesso ao exame de genotipagem ou em situações excepcionais de indisponibilidade temporária do exame, algumas recomendações devem ser observadas. Para essas situações, o quadro 3 sugere as alternativas de substituição para a escolha de um esquema ARV de resgate da primeira falha. Reforça-se mais uma vez, entretanto, que a utilização do teste de genotipagem deve ser a regra (BRASIL, 2007).
.jpg)
Fonte: (BRASIL, 2007)
Outros dois fármacos são recomendados como esquema de resgate: A enfuvirtida (IsF) e o Darunavir (IP). Porém existem alguns critérios para a utilização destes fármacos como esquema de resgate.
A enfuvirtida deve ser indicada somente, e principalmente, após se considerar os fatores clínicos e laboratoriais para estimar o risco de progressão da doença e morte na: a) indicação mais precoce, como por exemplo na doença clinica mais avançada , na imunodeficiência grave (CD4 < 100 cél./mm3) e disponibilidade de pelo menos uma outra droga com atividade na genotipagem ou; b) Postergar a indicação do novo esquema com enfuvirtida em pacientes com doença clinica menos avançada, melhor estado imunológico (CD4 > 100 cél./mm3) e indisponibilidade de droga com atividade detectada na genotipagem para compor o tratamento com o inibidor de fusão (BRASIL, 2007).
O darunavir deve ser indicado na composição de esquemas ARV de resgate quando: a) o teste de genotipagem ter sido realizado no Maximo 12 meses antes da troca para DRV; b) Haver ausência de outro IP/r com atividade plena, na ultima genotipagem; c) Conter atividade (I – intermediária ou S - plena) do DRV, na ultima genotipagem; d) houver a presença de uma droga ativa(I ou S) pertencente à outra classe, seja ITRN, ITRNN ou enfuvirtida (no caso de pacientes virgens dessa droga), na ultima genotipagem (BRASIL, 2007).
5 CONCLUSÃO
Nos últimos anos, o grande avanço verificado no campo da farmacologia dos anti-retrovirais (talvez a área de maior crescimento e desenvolvimento) mudou a vida das pessoas vivendo com AIDS, possibilitando um aumento da sobrevida e melhora da qualidade de vida. Apesar deste avanço, a facilidade com que os vírus adquirem resistência, torna imprescindível o desenvolvimento de novos medicamentos. Neste sentido, o Farmacêutico tem um papel fundamental seja no estudo direto de novos fármacos ou na atenção farmacêutica trabalhando a questão da adesão ao medicamento. A adesão ao medicamento é essencial para a manutenção da baixa carga viral e menor desenvolvimento de resistência. Através da genotipagem, hoje adotada apenas para pacientes com falha terapêutica, é possível uma terapia de resgate mais personalizada de acordo com o perfil de mutações apresentadas. A ampliação do arsenal terapêutico aliado a politerapia baseada na genotipagem para todos os pacientes com indicação de uso poderá quem sabe no futuro garantir o controle farmacológico desta infecção por períodos maiores, auxiliando no controle da epidemia.
REFERÊNCIAS
AFANI S., Alejandro; AYALA C., Marisol; MEYER K., Andrea; CABRERA C., Roy, ACEVEDO M., William. Resistencia primaria a terapia antirretroviral em pacientes com infección por VIH/SIDA en Chile. Rev Méd, Chile, v.133, n.3, p.295-301, mar.2005.
AHIJO, Bintou Ahmadou; VEALE, Rob; DUSÉ, Adriano G.; BECKER, Piet; MARAIS, Else. The nucleoside reverse trancriptase inhibitor didanosine, lamivudine, stavudine and zidovudine show little effect on the virulence of Candida albicans in vitro. International Journal of Antimicrobial Agents, Disponível on-line, v.32, n.1, p.186-191, Mar. 2008.
AIDS INFO. Disponível em - A Service of the US Departament of Health and Humans Services. Acesso em: 23 de Out. De 2008.
AIDS MAP. Disponível em < http://www.aidsmap.com/cms1032508.asp> - British HIV Association. Acesso em: 20 de Out. de 2008.
BIANCO, Rosana Del. Interação Medicamentosa para o Tratamento da Tuberculose e HIV. Tendências em HIV, São Paulo, v.2, n.1, p.14-17, 2007.
BISMARA, Beatriz Aparecida Passos. Padronização de Técnicas Moleculares para o Estudo da Resistência a Drogas Antiretrovirais em Crianças Infectadas pelo Vírus da Imunodeficiência Humana Tipo 1 (HIV-1) Via Perinatal. 2006. 166 f. Dissertação (Mestrado em Farmacologia) – Universidade Estadual de Campinas, Campinas.
BRASIL. Ministério da Saúde. Secretaria de Vigilância em Saúde. Programa Nacional de DST e AIDS. Boletim Epidemiológico – AIDS e DST Ano IV – nº 1 – 1ª a 26ª semanas epidemiológicas janeiro a junho de 2007.
BRITO, Ana Maria de; SZWARCHALD, Célia Landmann; CASTILHO, Euclides Ayres de. Fatores associados à interrupção de tratamento anti-retroviral em adultos com AIDS. Rio Grande do Norte, Brasil, 1999-2002. Rev Assoc Méd Brás, Rio Grande do Norte, v.52, n.2, p.86-92, 2006.
CARVALHO, Adelino Moreira de; SIMÕES, Ricardo Santos; OLIVEIRA, Fábio Hideo Martins; SIMÕES, Manuel de Jesus; OLIVEIRA FILHO, Ricardo Martins; NAKAMURA, Mary Uchiyama; KULAY Júnior, Luiz. Analise morfológica dos fígados e rins no binômio materno-fetal após tratamento de ratas prenhas com Ritonavir durante toda a prenhes. Rev Bras Ginecol Obstet, Minas Gerais, v.29, n.7, p.348-353, 2007.
CASEIRO, Marcos Montani; DIAZ, Ricardo Sobhie. Resistência Primária aos anti-retrovirais. Atividade Cientifica, São Paulo, Ano 3, Ed. 21, p.4-13, 2007.
CAVALCANTI, Ana Maria Salustiano. Resistência secundária aos antiretrovirais em indivíduos com AIDS e prevalência de sub-tipos do HIV-1 no nordeste do Brasil: 2002 a 2004. 2005. 75 f. Dissertação (Mestrado em Medicina Tropical) – Universidade Federal de Pernambuco, Recife.
CHI, Benjamin H.; CHINTU, Namwinga; CANTREL, Ronald A.; KANKASA, Chipepo; KRUSE, Gina; MBEWE, Felistas; SINKALA, Moses; SMITH, Peter J.; STRINGER, Elizabeth M.; STRINGER, Jeffrey S. A. Addition of Single-Dose Tenofovir and Emtricitabine to Intrapartum Nevirapine to Reduce Perinatal HIV Transmission. J Acquir Immune Defic Syndr, Disponivel On-line, v.48, n.2, p.220-223, jun. 2008.
COLOMBRINI, Maria Rosa Ceccato; LOPES, Maria Helena Baena de Moraes; FIGUEIREDO, Rosely Moralez de. Adesão à Terapia Antiretroviral para HIV/AIDS. Rev Esc Enferm USP, São Paulo, v.40, n.4, p.576-581, Dez.2006.
CORREIA, Cristina Maria Bacelar de Oliveira. Caracterização genética do vírus da imunodeficiência humana do tipo 1 (HIV-1), na zona de influencia do Hospital Geral de Santo Antonio, S.A. 2005. 92 f. Dissertação (Mestrado em Genética Molecular) – Departamento de Biologia, Universidade do Minho, Santo Antonio.
CUNTRELL, Amy; BROTHERS, Cindy; YEO, Jane; HERNANDEZ, Jamie; LAPIERRE, Didier. Abacavir and the potential risk of myocardial infarction. The Journal of Urology, on-line, v.371, n.9622, p.1413, apr.2008.
DEMETER, Lisa M.; DE-GRUTTOLA, Victor; LUSTGARTEN, Stephanie; BETTENDORF, Daniel; FISCHI, Margaret; ESHLEMAN, Susan; SPREEN, William; NGUYEN, Bach-Yen; KOVAL, Christine E.; ERON, Joseph J.; HAMMER, Scott; SQUIRES, Kathleen. Association os Efavirenz Hypersusceptibility with Virologic Response in ACTG 368, a Randomized Trial of Abacavir (ABC) in Combination with Efavirenz (EFV) and Indinavir (IDV) in HIV-infected Subjects with Prior Nucleoside Analog Experience. HIV Clinical Trials, New York, v.9, n.1, p.11-25, jan./feb. 2008.
DIAZ, Ricardo Sobhie. Orientações para o manuseio de testes de Resistencia anti-retroviral no paciente infectado pelo HIV. 1.ed. São Paulo: Abbott, 2006. p.115 e 128.
DOMINGOS, Hamilton. Efeitos Metabólicos Associados à Terapia Anti-retroviral Potente em Pacientes com AIDS. 2006. 103 f. Dissertação (Mestrado em Ciências da Saúde) – Convenio Rede Centro-Oeste UnB/UFG/UFMS, Campo Grande.
EIRA, Margareth da. Alterações na função renal em pacientes HIV/AIDS tratados com esquemas terapêuticos incluindo indinavir. 2004. 82 f. Dissertação (Mestrado em Ciências) – Universidade de São Paulo, São Paulo.
FALCI, Diego; BAY, Mônica; SPRINZ, Eduardo. Simplificação do Tratamento Anti-retroviral com a Utilização de Inibidor de Protease Reforçado com Baixas Doses de Ritonavir como Monoterapia de Manutenção em indivíduos HIV positivos com Supressão Viral. Tendências em HIV/AIDS, São Paulo, v.1, n.1, p.24-27, 2006.
FOCACCIA, Roberto; VERONESI, Ricardo. Tratado de infectologia. 3.ed. rev. e atual. São Paulo: Atheneu, 2007. p.111-113 e 238 -239.
GAFNI, Rachel I.; HAZRA, Rohan; REYNOLDS, James C.; MALDARELLI, Frank; TULLIO, Antonella N.; DE-CARLO, Ellen; WORRELL, Carol J.; FLAHERTY, John F.; YALE, Kitty; KEARNEY, Brian P.; ZEICHNER, Steven L. Tenofovir Disoproxil Fumarate and an Optimized Background Regimen of Antiretroviral Agents as Salvage Therapy: Impact on Mineral Density in HIV-Infected Children. Pediatrics, Illinois, v.118, n.3, p.710-719, sep. 2006.
GARFORTH, Scott J.; PARNIAK, Michael A.; PRASAD, Vinayaka R. Utilization of a Deoxynucleoside Diphosphate Substrate by HIV Reverse Transcriptase. Plos One, Peking, v.3, n.4, p.1-10, apr. 2008
HAZRA, Rohan; JANKELEVICH, Shirley; MACKALL, Crystal L.; AVILA, Nilo A.; WOLTERS, Pamela; CIVITELLO, Lucy; CHRISTENSEN, Barbara; JACOBSEN, Freda; STEINBERG, Seth M.; YARCHOAN, Robert. Immunologic, Virologic, and Neuropsychologic Responses in Human Immunodeficiency Vírus-Infected Children Receiving Their First Highly Active Antiretroviral Therapy Regimen. Viral mmunology, disponivel on-line, v.20, n.1, p.131-141, 2007.
HIRSCH, Martin S.; GÜNTHARD, Huldrych F.; SCHAPIRO, Jonathan M.; BRUN-VÉZINET, Françoise; CLOTET, Bonaventura; HAMMER, Scott M.; JOHNSON, Victoria A.; KURITZKENS, Daniel R.; MELLORS, John W.; PILLAY, Deenan; YENI, Patrick G.; JACOBSEN, Donna M.; RICHMAN, Douglas D. Antiretroviral Drug Resistance Testing in Adult HIV-1 Infection: 2008 Recommendations of an International AIDS Society-USA Panel. Clinical Infectious Diseases, Massachusetts, v.47, n.1, p.266-285, 2008.
HIV ROCHE. Disponível em < http://www.roche.pt/sida/tratamento/tratamento5.cfm> - Roche Farmacêutica Química. Acesso em: 20 de Out. de 2008.
HUGHES, Amelia; BARBER, Tristan; NELSON, Mark. New treatment options for HIV salvage patients: An overview of second generation Pis, NNRTIs, integrase innibitors and CCR5 antagonists. J Infect, London, p.1-10, mai. 2008.
ISAAKIDIS, Petros; RAGUENAUS, Marie-Eve; PHE, Thong; KHIN, Sam A.; KUOCH, Sokhan; KHEN, Sopheap; REID, Tony; ARNOULD, Line. Evaluation of a Systematic Substitution os Zidovudine for Stavudine-Based HAART in a Progran Setting in Rural Cambodia. J Acquir Immune Defic syndr, Disponivel on-line, v.49, n.1, p.48-54, sep. 2008
IUIS/WHO. ChemokineChemokine Receptor Nomenclature. Journal of Leukocyte Biology, Bethesda, v.70, n.1, p. 465-466, 2001.
KALMAR, Érika Maria do Nascimento. Avaliação da Resistência do HIV-1 às Drogas Anti-retrovirais em 150 Pacientes em Interrupção Terapêutica por mais de Seis Meses. 2007. 110 f. Tese (Doutorado em Medicina). Área de Concentração: Doenças Infecciosas e Parasitárias – Universidade de São Paulo, São Paulo.
KAPITSINOU, Pinelopi P.; ANSARI, Naheed. Acute renal failure in an AIDS patient on tenofovir> a case report. Journal of Medical Case Reports, Disponivel on-line, v.2, n.94, p.1-4, mar. 2008.
KATZUNG, Bertram G. Farmacologia: básica & clínica. 9.ed. Rio de Janeiro: Guanabara Koogan, 2006. p.677.
KAUL, Marcus; LIPTON, Stuart A. Mechanisms of Neuroimmunity and Neurodegeneration Associated with HIV-1 Infection and AIDS. J Neuroimmune Pharmacol, Disponivel on-line, v.1, n.1, p.138-151, fev. 2006
KLIMAS, Nancy; KONERU, Anne O’Brien; FLETCHER, Mary Ann. Overview of HIV. Psychomatic Medicine, Miami, v.70, n.5, p.523-530, 2008.
KONDO, William; ASTORI, Adriane de Assis Fischer; GOMES, Suria El-Kouba; FERNANDES, Rachelle de Brito; SASAKI, Maria das Graças; SBALQUEIRO, Renato Luiz. Avaliação dos efeitos colaterais da nevirapina em gestanets HIV positivo em Hospital Universitário do sul do Brasil. Rev Bras Ginecol Obstet, Curitiba, v.30, n.1, p.19-24, 2008
LAVRA, Zênia Maria Maciel. Obtenção Tecnológica de Antiretroviral Dose-Fixa Combinada à Base de Zidovudina, Lamivudina e Nevirapina. 2006. 88 f. Dissertação (Mestrado em Ciências farmacêuticas) – Universidade Federal de Pernambuco, Recife.
LEDERMAN, Michael M.; PENN-NICHOLSON, Adam; CHO, Michael; MOSIER, Donald. Biologia do CCR5 e seu Papel na Infecção e no Tratamento do HIV. JAMA, v.296, n.7, p.815-826, Ago.2006.
LOPES, Antonio Carlos. Diagnóstico e tratamento. V.3. São Paulo: Manole, 2007. p.1037 – 1040, 1045 – 1053 e 1056.
LORETE, Roberta dos Santos. Diversidade e resistencia do HIV-1 em gestantes soropositivas provenientes das regiões Sul e Sudeste do Brasil. 2005. 117 f. Dissertação (Mestrado em Biologia Celular e Molecular) – Instituto Oswaldo Cruz, Rio de Janeiro.
LOS ALAMOS. Disponivel em – HIV Laboratório Nacional de Los Alamos. Acesso em: 19 de Out. de 2008.
MACARTHUR, Rodger D.; NOVAK, Richard M. Maraviroc: The First of a New Class of Antiretroviral Agents. Clinical Infectious Diseases, Massachusetts, v.47, n.1, p.236-241, 2008.
MALLAL, Simon; PHILLIPS, Elizabeth; CAROSI, Giampiero; MOLINA, Jean-Michel; WORKMAN, Cassy; TOMAZIC, Janez; JABEL-GUEDES, Eva; RUGUNA, Sorin; KOZYREV, Oleg; CID, Juan Flores; HAY, Phillip; NOLAN, David; HUGHES, Sara; HUGHES, Arlene; RYAN, Susanna; FITCH, Nicholas; THORBORN, Daren; BENBOW, Alastair. HLA-B*5701 Screening for Hypersensitivity to Abacavir. N Engl J Med, Massachusetts, v.358, n.1, p.568-579, 2008
MANENTI, Sandra Aparecida. Epidemiologia e Caracterização Molecular do HIV-1 em Gestantes do Sul de Santa Catarina do Período de Janeiro a Dezembro de 2007. 2008. 136 f. Dissertação (Mestrado em Ciências da Saúde) – Universidade do Extremo Sul Catarinense, Criciúma.
MASHO, Saba Woldemichael; WANG, Cun-Lin; NIXON, Daniel E. Review of tenofovir-emtricitabine. Therapeutics and Clinical Risk Management, Virginia, v.3, n.6, p.1097-1104, 2007.
MATHIAS, Camila Fernandes Venneri; MATHIAS, Cícero Vanneri; SIMÕES, Ricardo Santos; OLIVEIRA-FILHO, Ricardo Martins; AMED, Abes Mahmed; SIMÕES, Manuel de Jesus; KULAY JÚNIOR, Luiz. Efeitos do Uso Crônico do nelfinavir sobre a prenhez da rata albina. Rev Bras Genecol Obtet, SãoPaulo, v.28, n.3, p.184-189, 2006
MELO, Ladjane Santos Wolmer. Características dos pacientes que evoluíram para óbito, em oito anos de terapia anti-retroviral potente, no Recife – Brasil. 2007. 69 f. Dissertação (Mestrado em Ciências da Saúde) – Universidade Federal de Pernambuco, Recife.
MONTEIRO, Vandessa Cristina da Silva; SENA FILHO, José Guedes da; MEDEIROS, Flavia Patrícia Morais de; ALBUQUERQUE, Miracy Muniz de; ROLIM, Pedro José. A Utilização do Mesilato de Nelfinavir na Terapia Anti-retroviral de Combate a AIDS. Informa, Recife, v.18, n.11. p.26-32, dez. 2006.
MORALES, Lydia G. Rivera; CRUZ, Itza E. Luna; ALFANO, Gerardo Ramos; SÁNCHEZ, Marco Ivan Ordaz; JIMENEZ, Javier Ramos; GUILLÉN, Paulo Lopez; GUERRA, Reyes Tamez; PADILLA, Cristina Rodriguez. Diverdidad Genética Del Vírus de Inmunodeficiencia Humana: Una Perspectiva General. Revista Salud Pública y Nutrición, Dicponivel on-line, v.6, n.1, p.1-9, jan./mar. 2005.
MOTA, Dunya Rodrigues; SIMÕES, Manuel de Jesus; AMED, Abes Mahmed; CARVALHO, Adelino Moreira de; OLIVERIA-FILHO, Ricardo Martins de; KULAY JÚNIOR, Luiz. Efeitos do Uso Crônico do Amprenavir sobre a Prenhez da Rata Albina. Rev Bras Ginecol Obstet, São Paulo, v.26, n.3, p.207-211, 2004.
MUNDERI, P.; KITYO, C.; WALKER, A. S.; GILKS, C.; GIBB, D. M.; SSALI, F.; BABIKER, A. G.; REID, A.; BRAY, D.; DARBYSHIRE, J. H.; GROSSKURTH, H.; MUGYENYI, P. Twenty-four-week safety and tolerability of nevirapine vs. abacavir in combination with zidovudine/lamivudine as first-line antiretroviral therapy: a randomized double-blind trial (NORA). Tropical Medicine and International Health, disponivel on-line, v.13, n.1, p.6-16, jan. 2008.
MURALHA, Acácio; REISNER, Márcio L.; CURI, André L. L. Retinopatia associada ao uso de didanosina. Arq Bras Oftalmol, rio de Janeiro, v.64, n.1, p.465-467, 2001.
PEÇANHA, Emerson Poley; ANTUNES, Octavio A. C.; TANURI, Amilcar. Estratégias Farmacológicas para a Terapia Anti-AIDS. Quim. Nova, Rio de Janeiro, v.25, n.6b, p.1108-1116, nov./dec.2002.
PEREIRA, J. Miguel Azevedo; PEREIRA, J. Moniz. Tropismo celular e patogênese da infecção pelo HIV-2. In: CONGRESSO VIRTUAL HIV/AIDS, 2., 2001, on-line. Anais eletrônicos... on-line: SIDAnet, 2001. Disponível em: Acesso em: 08 de Out. 2008.
QUEREDA, Carmen; CORRAL, Iñigo; MORENO, Ana; PÉREZ-ELÍAS, Maria J.; CASADO, José L.; DRONDA, Fernando; RODRIGUEZ-SAGRADO, Miguel A.; HERNÁNDES, Beatriz; MORENO, Santiago. Effect of Treatment with Efavirenz on Neuropsychiatric Adverse Events of Enterferon in HIV/HCV-Coinfection Patients. J Acquir Immune Defic Syndr, Madrid, v.49, n.1, p.61-63, sep. 2008.
RAMOS NETO, Saulo Salustiano. HIV E AIDS: Histórico, Patologia, Diagnóstico, Tratamento e Perspectivas sobre o mal do século. 2004. 81 f. Trabalho de Conclusão de Curso (Graduação em Farmácia) – Universidade do Extremo Sul Catarinense, Criciúma.
RANG, H. P.; DALE, M. Maureen. Farmacologia. 5.ed. Rio de Janeiro: Elsevier, 2005. p.74-+6 e 749-751.
RAVI, Punna Rao; KOTREKA, Udaya Kanth; SAHA, Ranendra, Narayan. Controlled Release Matrix Tablets of Zidovudine: Effect of Formulation Variables on the In Vitro Drug Release Kinetics. AAPS PharmSciTech, Disponivel on-line, v.9, n.1, p.302-313, jan./mar. 2008.
ROSEN, Sydney; LONG, Lawrence; FOX, Matthew; SANNE, Ian. Cost and Cost-Effectiveness of Switching From Stavudine to Tenofovir in First-Line Antiretroviral Regimens in South Africa. J Acquir Immune Defic Syndr, Disponivel on-line, v.0, n.0, p.1-11, jul. 2008.
SANTOS, Thiago C. A Prevalência de fatores de risco para baixa adesão na terapia com enfuvirtida, nos usuários soropositivos para o HIV em tratamento nos centos de referencia em Porto Alegre – RS. DST – J bras Doenças Sex Trasm, Porto alegre, v. 18, n.4, p.247-253, 2006.
SBI. Sociedade Brasileira de Infectologia. Disponível em < http://www.sbinfecto.org.br/default.asp?site_Acao=mostraPagina&paginaId=134&mNoti_Acao=mostraNoticia¬iciaId=356> - PEC Programa de Educação Continuada. Acesso em: 20 de Out. de 2008.
SCHNEIDER, Serge; PELTIER, Alexandra; GRAS, Alain; ARENDT, Vic; KARASI-OMES, Christine; MUJAWAMARIWA, Anastasie. Efavirenz in Human Breast Milk, Mothers’, and Newborns’ Plasma. J Acquir Immune Defic Syndr, Disponível on-line, v.48, n.4, p.450-454, ago. 2008.
SILVEIRA, Alexsander Augusto da. Caracterização da Diversidade Genética do HIV-1 em Pacientes do Estado do Mato Grosso do Sul. 2007. 84f. Dissertação (Mestrado na área de concentração de Imunologia) – Universidade Federal de Goiás, Goiânia.
SINAN. Ministério da Saúde. Doenças de agravo. Disponível em: HTTP://dtr2004.saude.gov.br/sinanweb/novo/. Acesso em: 03 de dez. 2008.
SOUZA, Marcus Vinícius Nora de, ALMEIDA, Mauro Vieira de. Drogas Anti-VHI: Passado, Presente e perspectivas futuras. Quim. Nova, Rio de Janeiro, v.26, n.3, p.366-372, mai./jun.2003.
SOUZA, Marcus Vinicius Nora de. Fármacos Inibidores de Fusão: uma Nova Estratégia no Combate à Replicação do Vírus VIH. Acta Farm. Bonaerense, Rio de Janeiro, v.24, n.2, p.291-299, 2005.
STRINGER, James R.; BEARD, Charles B.; MILLER, Robert F.; WAKEFIELS, Ann E. A New Name (Pneumocystis jiroveci) for Pneumocystis from Humans. Emerging Infectious Diseases, Atlanta, v.8, n.9, p.891-896, sep. 2002.
STROHL, William A.; ROUSE, Harriet; FISHER, Bruce D. Microbiologia ilustrada. Porto Alegre: Artmed, 2004. p.374-376, 378 e 387-389.
TENORE, Simone de Barros; FERREIRA, Paulo Roberto Abrão. Uso da Enfuvirtida no Tratamento de Pacientes com Falha Virológica aos Anti-retrovirais. Tendências em HIV/AIDS, São Paulo, v.2, n.1, p.23-27, 2007.
THE BIOLOGY PROJECT. The Replication Cycle of HIV, 2000. Disponível em: < http://www.biology.arizona.edu/immunology/tutorials/AIDS/treatment.html> Acesso em: 13 de Outrubro de 2008.
TORRES, Harry A; ARDUINO, Roberto C. Fosamprenavir Cálcio e ritonavir na infecção pelo HIV. Expert Rev Anti Infect. Ther, Texas, v.5, n.3, p.349-363, 2007.
TORTORA, Gerard J.; FUNKE, Berdell R.; CASE, Christine L. Microbiologia. 8.ed. Porto Alegre: Artmed, 2005. p.546-547.
UNAIDS/WHO, Joint United Nations Programme on HIV/AIDS/World Health Organization, AIDS, epidemic update: December 2007, eneva, 2007. Disponivel em .
WYL, Viktor Von; YERLY, Sabine; BÖNI, Jürg; BÜRGISSER, Philippe; KLIMKAIT, Thomas; BATTEGAY, Manuel; NERNASCONI, Enos; CAVASSINI, Matthias; FURRER, Hansjakob; HIRSCHEL, Bernard; VERNAZZA, Pietro L.; RICKENBACH, Martin; LEDERGERBER, Bruno; GÜNTHARD, Huldrych F. Factors Associated with the emergence of K65R in Patients with HIV-1 Infection Treated with Combination Antiretroviral Therapy Containing Tenofovir. Clinical Infectious Diseases, Massachusetts, v.46, n.1, p.1299-1309, 2008.
Publicado por: Tago Rodrigues
O texto publicado foi encaminhado por um usuário do site por meio do canal colaborativo Monografias. Brasil Escola não se responsabiliza pelo conteúdo do artigo publicado, que é de total responsabilidade do autor . Para acessar os textos produzidos pelo site, acesse: https://www.brasilescola.com.























