Sumário
1. RESUMO
Devido às descobertas da ciência, foi possível obter uma correlação e envolvimento do mecanismo genético. Conseguindo assim, mapear o gene que acomete essa patologia conforme os critérios estabelecidos pelo National Institute of Health (NIH). A neurofibromatose tipo 1 (NF1), é uma patologia autossômica dominante, com estimativa de incidência de 1:3.000 nascidos vivos. Observando muitos preconceitos em torno da NF1, que acomete crianças, adolescentes e suas respectivas famílias na vida escolar e social. Esta patologia caracteriza-se principalmente por possuir uma grande diversidade de apresentações clínicas e tendo a mutação no cromossomo 17q.11.2 do genoma humano. Os portadores apresentam manchas café com leite (MCCL), nódulos de Lisch e neurofibromas plexiformes ou cutâneos e gliomas. Entretanto o objetivo deste trabalho é desmistificar a NF1, auxiliar com esclarecimentos da fisiopatologia, diagnóstico características e seus diversos tratamentos da doença, bem como para os demais profissionais.
ABSTRACT
Due to the discoveries of science, it was possible to obtain a correlation and genetic mechanism involved. Thus, mapping the gene that affects this pathology as the criteria established by the National Institute of Health (NIH). Neurofibromatosis Type 1 (NF1), is autosomal dominant, with estimated incidence of 1:3,000 live births. Noting many preconceptions around the NF1, which affects children, adolescents and their families in school and social life. This pathology is characterized primarily by possessing a wide range of clinical presentations and having the mutation on chromosome 17q 11.2 of the human genome. The feature café-au-Lait Spot (MCCL), Lisch nodules and neurofibromas plexiformes or cutaneous and gliomas. However, the aim of this paper is to demystify the NF1, assist with clarification of pathophysiology, diagnosis, characteristics and its various treatments of the disease, as well as for other professionals.
Key-words: Diagnostics; Neurofibromatosis (NF); Genes; Incidence factors; Institutes.
2. INTRODUÇÃO
Diante da dificuldade de compreensão desta patologia tanto pela população e até mesmo para os profissionais da saúde, a Neurofibromatose tipo 1 (NF1) ou doença de Friedrich Daniel Von Recklinghausen na sociedade é uma grande diversidade de apresentações clínicas e com suas especificidades em cada caso. Com uma alta ocorrência de 1:3.000 nascidos vivos, como retratam alguns estudos que decorrem desta patologia. Assim esclarecer a importância dada ao aconselhamento genético na qual analisaria o cromossomo 17q11.2 dos acometidos pela doença (AMANF, 2008). A Neurofibromatose é uma desordem genética que abrange três principais doenças, como a NF1, Neurofibromatose tipo 2 (NF2) e Schwannomatose (SCH). Cada uma com as suas características, especificidades e clinicamente distintas. Pautando desta descrição e conforme a fundamentação proposta no trabalho, a NF1 se define como uma patologia genética de caráter autossômico dominante, com uma vasta sintomatologia, prognóstico e apresentações clínicas, conforme os critérios estabelecidos pelo National Institute of Health (NIH), realizada em Bethesda-EUA, em 1988 (GUTMANN, 2012).
As principais manifestações dessa patologia são manchas café com leite, nódulos de Lisch e neurofibromas, margeando 90% dos pacientes até a puberdade. Será um trabalho de grandes referências para os que se interessam pela NF1, contudo, se divulga pouco sobre o assunto, fazendo com que a doença seja negligenciada em muitos pontos. Entretanto o objetivo deste trabalho é desmistificar a NF1, auxiliar com esclarecimentos de diagnósticos, características e seus diversos tratamentos da doença, bem como para os demais profissionais. Observando muitos preconceitos em torno da NF1, que acomete crianças, adolescentes e suas respectivas famílias na vida escolar e social (KORF, 1992).
Uma justificativa seria que diante da invisibilidade a qual essa doença apresenta, é de suma importância esclarecer aspectos correlacionados à NF1 para com a sociedade, pois quase sua totalidade é leiga no assunto. Considerando especialmente o fato dela se tratar de uma desordem genética, tanto o paciente quanto os familiares avaliaram as implicações e falam que tem necessidade de um acompanhamento de múltiplos profissionais como dermatologia, psicólogo, neurologia entre outros (AMANF, 2008). Seu fundamento e que a Neurofibromatose
(NF) é uma doença de caráter desconhecido na sociedade, todavia sendo estudada por grandes pesquisadores há muito tempo. Todavia, descrevem-se também áreas pigmentadas na pele, mas, aparentemente, não incluíam as manchas café com leite como parte da doença, ressaltando-se que raramente os tumores de sistema nervoso central são acompanhados das manifestações cutâneas e que nenhum dos pacientes apresentava manifestações neurológicas, exceto baixo nível de inteligência (CANALE, 1972).
Contudo a NF1, é uma desordem genética, com prevalência relevante na sociedade, margeando em torno de 50% das crianças uma vez que estas herdam de seus pais e os outros 50%, sofrem mutação espontânea, ou seja, os pais são sadios. Entretanto, essa patologia é de caráter autossômico dominante e constitui um verdadeiro desafio para diversas especialidades médicas, por acometer vários locais no corpo, como por exemplo, pele, órgãos, olhos, causando também lesões ósseas e o sistema nervoso central. Pautando-se nesse entrave, existem vários tipos de características para afirmar o diagnóstico desta doença. Estudos apontaram que 70% dos pacientes com a NF1, podem ser diagnosticados antes de 1 ano de idade. Contudo, como ainda existem pessoas que ainda são diagnosticadas nesta fase sabendo que a manifestação da patologia ocorre antes de um ano de idade?
Artigos selecionados conforme a qualidade de relevância com o tema proposto. Revistas de Ciências Médicas e Biológicas Google Acadêmico, Livro NEUROFIBROMATOSE clínica, Genética e Terapêutica (Mauro Geller; Agnaldo Bonalumi Filho), PubMed- NCBI National Library of Medicine e Scientific Eletronic Library Online SCIELO. A procura dos artigos foi limitada entre os anos de 1981 e 2017, usados como descritores Neurofibromatose (NF), Diagnósticos, Genes, Epidemiologia, Fatores de incidências e Institutos.
3. REVISÃO DA HISTÓRIA LITERÁRIA E SEU ACOMETIMENTO GENÉTICO DA NF1 NA SOCIEDADE
A Neurofibromatose (NF) é uma doença de caráter desconhecido na sociedade, todavia sendo estudada por grandes pesquisadores há muito tempo. Pautando nesta informação, segundo o autor Mark Akensidi (1785, p 344) “apresentaram relatos maiores referentes a um paciente apelidado de homem verruga Wart Man relatado por Tilesius Von Tileau em 1793 com nódulos cutâneos, manchas nas pernas, escaras, prurido e uma enorme cabeça” (RICCARDI, 1992). Logo depois Robert Smith (1849), relatou dois casos de pacientes com neuromas, semelhantes àqueles descritos posteriormente por Friederich Daniel Von Recklinghausen (1882) (SMITH,1963).
O médico pesquisador Von Recklinghausen, ao reconhecer a NF como entidade patológica descreveu dois casos de NF múltipla, um dos quais foi à necropsia. Postulou que os tumores ao longo dos grandes nervos periféricos e também os falsos neuromas da pele, provinham do tecido conjuntivo da bainha dos nervos e dos plexos nervosos, particularmente da camada de tecido conjuntivo que envolve um feixe de fibras nervosas de um nervo periférico e do tecido conjuntivo que envolve cada fibra nervosa no interior de um feixe nervoso.
Todavia, descrevem-se também áreas pigmentadas na pele, mas, aparentemente, não incluía as manchas café com leite como parte da doença, ressaltando-se que raramente os tumores de sistema nervoso central são acompanhados das manifestações cutâneas e que nenhum dos pacientes apresentava manifestações neurológicas, exceto baixo nível de inteligência (CANALE, 1972). De acordo com Verocay (1910) propôs o nome de neurinoma a esses tumores nervosos e postulou sua origem a partir de células indefinidas (neuroectodérmicas), capazes de dar início aos vários tipos de tumores da doença de Von Recklinghausen.
Presiser e Davenport (1918) “afirmaram que a doença NF não está ligada ao sexo, obedecendo assim à lei de Mendel como um caráter dominante” (CANALE, BEBIN, 1972). Contudo, em 1937 obteve-se outra manifestação clínica da NF: nódulos na superfície da íris, com elevações de aspecto gelatinoso e de forma arredondada, caracteristicamente numerosos, de natureza hamartomatosas variando
em coloração, de transparentes ao amarelo/marrom, bilaterais e bem definidos, recebendo o nome de nódulos de Lisch (RICCARDI, 1992). Entretendo em 1918, estabeleceu-se que a doença não fosse ligada ao sexo da pessoa e sim obedecendo a um estudo lógico sobre a lei de Mendel, que nos propunha pensar que a doença seria de caráter autossômico dominante.
Dois relatos clássicos ocorreram com portadores distintos, que devido às características apresentadas, foi erroneamente descrito como portador da NF. Pois apresentavam alterações nos membros superiores, nódulos cutâneos, manchas nas pernas, escaras e prurido, denominado como “Homem Verruga ou Wart-Man” pelo pesquisador Tilesius Von Tileau em 1793 e “Homem Elefante” pelo pesquisador Joseph Carrey Merrick no século XIX. Devido a essa patologia, eram submetidos a exposições em espetáculos circenses (CANALE, 1972). Apesar das diversas manifestações clínicas, com o avanço da ciência, foi possível descobrir a correlação e envolvimento do mecanismo genético, assim conseguindo mapear o gene que acomete essa patologia conforme os critérios estabelecidos pelo NIH, realizada em Bethesda-EUA, em 1988. (DARRIGO, 2008).
Surgiram descrições de uma forma central de NF, que idealiza uma origem nervosa dos tumores descrita nas famílias, com vários portadores afetados como salienta o autor Huson. Conforme esse primeiro caso, em 1882 o autor Friederich Daniel Von Recklinghausen, informou esta patologia de forma correta e mais eficaz, que pudesse diagnosticar os neurofibromas e a origem nervosa dos tumores. Que constituiu um marco importante na evolução mundial sobre o assunto que caracterizaria a patologia em 1896 conforme o acometimento genético ao paciente (SOUZA, 2009).
Todavia os autores Rasmussen, Yang e Friedman (2001) concluem, ao estudar o perfil da mortalidade na NF1, que os portadores da doença apresentam menor expectativa de vida em relação à população em geral. Além disso, tumores cerebrais e tumores de tecido conectivo e de partes moles são as causas de óbito mais frequentes. Entretanto a elevação da incidência de desordens de hiperatividade e déficit de atenção em crianças com NF1, foi relatado pelos autores Mautner et al. (2002) possui uma associação entre essas desordens e os problemas sociais de aprendizado presentes nos portadores desta doença (MAUTNER et al., 2002). Como se pode observar a um grande estudo margeando 10 anos de buscas para a cura aos portadores conforme a característica do acometimento genético.
3.1. Fatores que levam aos acometimentos genéticos
A desordem genética é correlacionada a NF2, devido às modificações que acontecem no braço longo do cromossomo. Todavia o acometimento, mais raro na população é a desordem pela NF2, tendo um grupo de nascidos vivos 1:40.000, entretanto a quantidade genômica é de 110 Kb e 17 éxons acometida no cromossomo 22q12.1 (BARKER et al., 1987).
Contudo estudos na literatura abordam que a NF1 é uma desordem genética dominante acometida no cromossomo 17 do braço longo 17q11.2, tornando-a persuasiva às variações nas apresentações clínicas na população. Pautando nessa dominância, os nascidos vivos são aproximadamente 1:3.000, pessoas afetadas pela doença podem ser de herança genética equivalente a 50% e os outros 50 % de desenvolvimento espontâneo, podendo passar essa variação até a sexta geração na família (DARRIGO, 2008). Tendo em vista a quantidade genômica de 350 Kb de DNA e 51 éxons como se observa na figura 1, já os duplicados primários, sofrem “splicings” de opções nos éxons 9a, 23a e 48a. Segundo Ferreira (1993) “quando a NF1 é correlacionada a outras doenças que acometem a população tal como fibrose cística, Diatetes mellitus tipo 1 gira em torno de 1:13.000 nascidos vivos. Tornando-a foco principal do trabalho bibliográfico.
Figura 1 – Figura 1. Comparação cromossômica da doença NF1 e tipo 2, os com o auxílio da seta (amarela) se localiza o gene afetado pela doença.
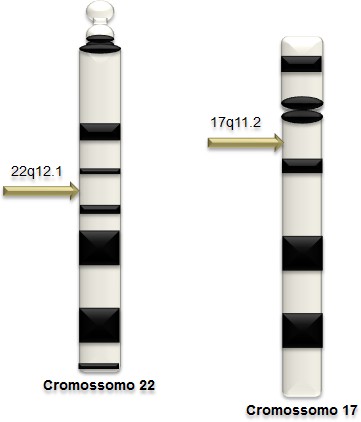
Fonte: Adaptado do Site Defectos al Nacimiento –(Ano 2013) Link acesso: http://infogen.org.mx/sindrome-de-delecion-del-cromosoma-22q11-2-tambien-llamado-sindrome-de- digeorge-y-sindrome-velo-cardio-facial/. Site: Cancro na Família – (Ano 2012) Link acesso: http://www.cancronafamilia.com/pt/cancro-da-mama/risco-na-familia/cancro-hereditario/genes- brca/risco-em-brca1/.
A isoforma tipo 1, demonstra no SNC uma dominância no cérebro, não barra os aminoácidos codificados pelos éxons 9a, 23a e 48a. A isoforma tipo 2, existente nas células de SCH, é um representativo do éxon 23a. A isoforma tipo 3, é um decorrente do “splicing” do éxon 48a, contudo a isoforma tipo 4, são dos éxons 23a e 48a. Todavia, as duas últimas isoformas manifestam-se na musculatura cardíaca, esquelética e lisa sendo, a expressão mais comum, que codifica a proteína com 2818 resíduos de aminoácidos de 327 kDa (GELLER, 2004).
A região conhecida das funções, apresenta um domínio catalítico da proteína ativadora de GTPase Ras-específica (domínio GAP) que contém 300 aminoácidos (XU et al., 1990). Partindo deste princípio, a neurofibromina, é uma proteína citoplasmática, que regula de forma negativa a função da proteína Kinase (Ras p21) proteína quinase ativada por mitógeno (MAPK) que provem a sobrevivência e a proliferação celular (JEAN, 2015). Quando a estrutura da proteína ativadora GTPase Ras-específica (Domínio GAP) se choca com Ras, ocorre uma modificação conformacional e a atividade GTPásica inerente e fortemente estimulada. Por tanto, a GTP ligado a Ras (na forma ativa) é imediatamente hidrolisado a GDP (na forma inativa), consequentemente a via de sinalização mitogênica celular é diminuída.
Então podemos falar que a neurofibromina aponta a diminuição da sinalização da manifestação celular, ou seja, regula negativamente a via de transdução de sinal relacionada por Ras (DAVID, 2012).
Embora a cascata Ras não consiga controlar o crescimento e diferenciação celular, então conclui que a neurofibromina não funcional, resulta em ativação constitutiva (não controlada) dessa via central de sinalização e crescimento celular (HANNAN, 2006). Estudos literários relataram outra proteína dando início para o desenvolvimento do isolamento, clonagem e caracterização em 1990 pelos autores Cawthon et al., Viskochil et al. e Wallace et al. onde definiu-se que a transformação maligna da NF1 que associa-se, pela perda de expressão da proteína p16, é secundária à deleção do gene CDKN2A/p16, desenvolvendo a inativação deste gene que ocorre durante a transformação maligna dos neurofibromas (NIELSEN, 1999).
O coma mixedematoso (CM) é uma emergência endocrinológica rara, porém letal e consiste na expressão extrema do hipotireoidismo, tem que fazer o tratamento, pois sem ser feito pode agravar cada vez mais. Antigamente por não haver tratamento 80% das pessoas que possuía a doença morriam hoje em dia com o tratamento a chance de mortalidade cai pra 20%.
Embora a patologia seja autossômica dominante. O gene da NF1 foi mapeado e clonado na região pericentromérica do cromossomo 17q11.2 (12). A neurofibromina é um produto do gene que apresenta atividade de uma proteína ativadora da GTPase e é capaz de fazer a regulação negativa da proto-oncogene p21-ras. A perda de sua função pode levar ao crescimento celular descontrolado e à formação de tumores, daí sua conhecida associação com tumores benignos e malignos.
Além disso, a produção anormal de neurofibromina suprime a expressão de fas-ligante, o que pode evitar a apoptose das células T CD4+, importante no desenvolvimento da autoimunidade (13). Ainda que menos frequente do que sua associação com tumores, a NF1 tem sido associada a doenças autoimunes, tais como: esclerose múltipla, esclerose sistêmica, lúpus eritematoso sistêmico, glomerulonefrite membranosa etc. No entanto, há poucos casos relatados de associação de NF1 com doença autoimune tireoidiana (SCIELO,2013).
Mulheres com NF1 que possibilitam a gravidez, frequentemente se perguntam se os sintomas aumentarem ou até mesmo quem possui a doença e não sabe na gravidez pode vim a aparecer os primeiros sintomas, como os neurofibromas, que pode aumentar ou aparecem pela primeira vez durante a gravidez. O risco de desenvolvimento de novos neurofibromas durante a gravidez pode estar acima de 50%. Neurofibromas que crescem mais durante a gravidez podem, em alguns casos, diminuir de tamanho após o parto. Isso é mais provável por uma função de edema geral e acúmulo de líquidos, que naturalmente ocorre durante a gravidez (Rev Bras Clin Med,2008).
4. DESCRIÇÃO FISIOPATOLÓGICA E DIAGNÓSTICOS EXISTENTES
Os diferentes diagnósticos clínicos são realizados na infância conforme critérios clínicos estabelecidos pelo NIH. O diagnóstico precoce permite proporcionar aos pacientes e familiares um conforto psicossocial diante de tamanha complicação que esta patologia causa se não diagnosticada a tempo. Estudos apontaram que 70% dos pacientes com a NF1, podem ser diagnosticados antes de um ano de idade. Existem casos que a herança é herdada de teus genitores, que podem ser diagnosticados nos primeiros anos de vida e antecedentes familiares (DEBELLA, 2000).
No entanto, a prevalência é relevante na sociedade, margeando em torno de 50% das crianças uma vez que estas herdam de seus pais e os outros 50%, sofrem mutação espontânea, ou seja, os pais são sadios. Alguns casos são inesperados como as efélides (sardas), neurofibromas e os nódulos de Lisch, que aparecem no decorrer da idade (KORF, 1992). Margeando os quatro anos de idade o diagnóstico poderá ser feito conforme NIH, porém os critérios estabelecidos podem ser imprecisos, devido aos casos inesperados, assim dificultando o diagnóstico tão preciso para o tratamento precoce. Como por exemplo, casos que só manifesta MCCL sem outras alterações, embora seja estridente o gene da NF1, as características clínicas dessa patologia são surpreendentemente variáveis (Tabela 2).
Tabela 2- Principais características clínicas acometidas pela patologia NF1 seguindo os critérios estabelecidos pela NIH
|
Sintomas |
Idade |
Incidência |
Característica |
|
Efélides (sardas) |
0 – 6 anos |
90% |
Região Axilar ou Inguinal |
|
Glioma |
1 ano 3 anos |
1 % 4 % |
Óptico |
|
Lesão Óssea |
Maioria crianças |
5% |
Pseudoartrose de um osso longo e Displasia da asa do esfenoide |
|
MCCL |
Todas as idades |
99% |
≥ 6 com diâmetro de 5mm, em pré- púberes Diâmetro de 15mm, em pós- púberes |
|
Neurofibromas |
10 anos 20 anos |
48% 84% |
≥ 2 Dois Cutâneos ou Plexiformes |
|
Nódulos de Lisch |
10 anos |
70% |
≥ 2 Hamartoma pigmentado na Íris |
Fonte: AdaptadoStumpf da, alksne jf, annegers jf. Neurofibromatosis: conference statement. Arch Neurol (Ano 1988).
Existem testes moleculares que ajudam em torno de 97% das mutações os pacientes/portadores nos critérios do diagnóstico clínico quando o mesmo é erroneamente diagnosticado. Vale ressaltar que este teste não prediz o grau de acometimento da patologia ou início para com a pessoa, ou até mesmo se o teste der negativo, não significara a ausência de uma mutação (MESSIAEN, 2003).
Para poder confirmar o diagnóstico, o paciente/portador precisará apresentar pelo menos dois ou mais aspectos indicados na tabela 2. Os exames complementares para diagnosticar a patologia são utilizada entre, tomografia computadorizada, a angiografia ou angiograma (injeção de contraste radiopaco (tintura) intravascular), colonoscopia (exame da porção superior do reto) e broncoscopia (exame óptico dos brônquios), ressonância magnética de imagem, espectroscopia de prótons (avaliação metabólica cerebral), exames cutâneos da hiper pigmentação com lâmpada “wood", exame oftalmológico, biópsia dos neurofibromas, avaliação auditiva, teste de QI e análise psicológica (FERNER RE, 2013).
Contudo, testes moleculares para o mapeamento da mutação do gene NF1, são efetuados principalmente em grávidas geralmente cerca de 5 a 10% sofrem delações do gene NF1, foram observados utilizando testes de Hibridização “in situ” por Fluorescência (FISH), análise polimorfismo conformacional de cadeia simples (SSCP) e de Heteroduplex, eletroforese em gel com gradiente de temperatura (TGDGE) ou desnaturante (DGGE) e o protein trucation test (PTT). O teste de proteína foi considerado um dos melhores testes por conseguir detectar as proteínas truncadas, entretanto é mais eficaz identificar quando são geradas “in vitro” (DUNNEN, OMMEN, 1999). Portanto, é possível analisar e identificar proteínas truncadas originadas de alelos onde as mutações produziram um códon de terminação. O método FISH é eficaz quando ocorre deleção total do gene em questão, ou seja, o mesmo não se hibridiza com a sonda fluorescente (FERNER RE, 2013). No próximo subcapitulo abordaremos sobre os tratamentos e Institutos de apoio aos portadores e familiares dessa patologia, de uma forma breve.
Com base na pesquisa clínica francesa do Pennsylvania Human Relations Commission (PHRC) NF1, coordenador Professor Pierre Wolkenstein, Henri Mondor Hospital, Cre’teil, França, possuiu 1083 pessoas que preenche os critérios dessa patologia. Contudo essa pesquisa foi colhida 30 amostra de sangue de pacientes
que possui a NF1 e através da análise da bioinformática sequenciamento de nova geração (NGS), observou-se o alinhamento da sequência foi realizado com o Ion Torrent Suite no Ion Torrent Browser Life Technologies (SABBAGH et al., 2013).
Os polimorfismos de um único nucleotídeo (SNPs) e as inserções curtas e / ou chamadas de deleções foram realizados usando o plugin Variant Caller no Ion Torrent Browser e as seqüências de DNA visualizadas usando o Integrative Genomics Viewer (IGV) do Broad Institute (Cambridge, MA, EUA). O software NextGENe v2.3.3 (Softgenetics, State College, PA, EUA) também foi usado para chamadas de SNPs e curtas inserções e / ou deleções, e sua visualização e anotação. Em resumo, os principais parâmetros de chamada foram escolhidos da seguinte forma: frequência alélica mínima (MAF) ≥ 10% para SNPs e inserções curtas e / ou deleções, profundidade mínima de seqüenciamento ≥6X para SNPs e ≥15X para inserções curtas e / ou deleções.
O filtro de corte de 10% MAF permitiu evitar qualquer resultado falso negativo. As variantes selecionadas usando esse filtro foram então analisadas em nosso fluxo de trabalho bioinformático, incluindo dois parâmetros principais para variantes: a pontuação variante (do software NextGENe, Softgenetics) e a recorrência da variante. O software NextGENe (Softgenetics) calculou uma pontuação variante para fornecer uma estimativa empírica da probabilidade de uma determinada variação não ser um artefato de sequenciamento ou erro de alinhamento. Esta pontuação dependia principalmente da cobertura da variante. À medida que as leituras perto do extremo 5 'são mais precisas do que leem no final 3', as correspondências que foram encontradas no início de uma contagem de leitura foram ponderadas mais fortemente do que as incompatibilidades que foram encontradas no final 3 'da leitura (SABBAGH et al., 2013).
Assim foi modulada por (i) a proporção do número de leituras para frente ao número de leituras reversas na localização da variante, (ii) frequências variantes nas direções direta e reversa, e (iii) o número de desajustes ocorridos em uma porcentagem mínima de leituras na região de 10 pb que é encontrada em qualquer lado da variante. Para uma série de 48 amostras, a chamada variante foi realizada para cada amostra separadamente. Todas as variantes foram então coletadas e agregadas. Como nenhum “hotspot” de mutação foi descrito, a chamada variante foi realizada para cada amostra separadamente. Todas as variantes foram então coletadas e agregadas. Como nenhum hotspot de mutação foi descrito no gene NF1, a presença de uma variante em mais de 2/48 amostras (de 48 pacientes não relacionados) provavelmente indicará um erro sistemático de sequenciamento ou polimorfismo (PASMANT et al., 2015).
Alguns exames complementares podem ser úteis para a avaliação da NF1 e são indicados somente para a avaliação de queixas específicas: Radiografia as alterações radiológicas encontradas com maior frequência são escoliose, cifose, cifoescoliose, áreas de erosão óssea, crescimento anormal do osso, pseudoartrose, meningocele, deformidade da parede posterior dos corpos vertebrais, peito escavado, lesões osteolíticas nos ossos longos, alterações do sistema nervoso central e displasia óssea do esfenoide, Tomografia computadorizada (TC) devido à diversidade das aparências radiológicas dos neurofibromas orbitários isolados, o uso combinado de TC de alta resolução e de ressonância nuclear magnética pode ser benéfico no pré-operatório desses tumores, ressonância nuclear magnética (RNM) a maioria dos tumores identificados são astrocitomas de grau I, pois os tumores cerebrais ocorrem com uma frequência aumentada e são exclusivamente astrocíticos e 15% dos pacientes apresentam Glioma ópticos, Eletroencefalograma em pacientes com NF1 apresentam prevalência de 4,2% de crises epilépticas, o dobro da população no geral têm o potencial evocado juntamente com a RNM, pode ser utilizado para a detecção e monitoramento de complicações cerebrais em pacientes com NF1; Eletroneuromiografia estuda a velocidade de condução neuromuscular. Os pacientes com tumores dos nervos periféricos, geralmente isolados, apresentam uma variedade de sinais e sintomas que podem estar presentes na NF1, embora apresente múltiplas massas e envolvendo vários nervos; Exame com lâmpada de fenda é utilizado para a detecção dos nódulos de Lisch (Rev Bras Clin Med,2008).
5. TRATAMENTOS E INSTITUTOS DE APOIO AOS PORTADORES E FAMILIARES
Ainda não existe uma cura para esta patologia, porém a tratamentos que ameniza os sintomas. Estudos mostraram em cinco meninas e treze meninos que eram portadores da NF1 e apresentavam Glioma óptico (nervo que liga a visão), tiveram uma regressão espontânea de massas hipotalâmicas e de quiasma óptico. Das dezoito crianças, duas mostraram massa sugestiva de neoplasia, no exame de ressonância magnética, que é o exame utilizado para descobrir se a pessoa tem o glioma, e foi verificado que ambos, a visão e endócrina não foram alterados (NEURINOMA, 2003).
Entretanto as MCCL são normais o aparecimento nessa patologia, algumas pessoas se incomodam com as manchas, e levam a realizarem tratamentos a laser que são específicos. Mas para as manchas que são características da patológica, este método, apenas ajudara a amenizar e ajudar os portadores a terem uma qualidade de vida preferível. Pois não acontecera o desaparecimento das mesmas na pele dessas pessoas (AMANF, 2008).
Os neurofibromas que é outra característica poderão ser removidos através de cirurgias, porém não tem uma garantia que ele não crescerá novamente, como por exemplo, um caso de um rapaz de vinte seis anos que dês dos três apresentavam nódulos pequenos, mais um desses nódulos no abdômen teve um crescimento acelerado e ele sentia muita dor, e com isso o médico optou pela cirurgia Os plexiformes que são um tipo de neurofibromas que são mais resistentes, eles crescem em torno dos olhos, no braço, pernas e troncos. O meio de tratamento é a cirurgia, entretanto não é eficaz por que a maioria dos plexiformes não consegue retirar a raiz por completo, e com isso acaba tendo a possibilidade do crescimento novamente (REVISTABRASIL, 2012).
A patologia afeta também o aprendizado, mas a forma como será afetado dependerá do metabolismo de cada pessoa, contribuindo assim para o crescimento e influenciando no atraso da puberdade. Alguns médicos indicam o tratamento com hormônios para estimular o crescimento e a vinda da puberdade. Estudos mais recentes mostram tratamento com retinóides (ácidos graxos aromáticos), potenciais candidatos para testes clínicos em neurofibromas plexiformes associados a NF1 e
retinóides, o ácido retinóico 13 -cis (Accutane), estão disponíveis como drogas orais, o que pode facilitar sua administração. 4-Fenilbutirato (4-PBA) testado para a infusão intravenosa e necessita administração através de uma bomba portátil para injeção contínua devido à meia vida curta da droga no sangue. Uma preparação oral de PBA também está sendo testado em um teste clínico tratando tumores cerebrais malignos; esta pode ser uma alternativa à versão intravenosa (AMANF, 2008).
Entre vários Institutos e associações de apoios existentes no Brasil e no mundo, informaremos uma associação (AMANF) Associação Mineira de Apoio aos Portadores de Neurofibromatose em Belo Horizonte e outras cidades de Minas Gerais que foi criada e 2002 a partir de portadores da NF1 e seus familiares. Os participantes são os próprios portadores, a família, amigos e os profissionais da área de saúde. A associação tem como objetivo apoiar, discutir, trocar ideias, relatos e experiências sobre essa patologia. A partir de março de 2005 os pacientes passaram a contar com o Centro de Referência em Neurofibromatose de Minas Gerais, onde podem ser atendidos gratuitamente e receber todas as informações médicas disponíveis e orientações sobre a NF. Atualmente, o Centro de Referência conta também com atendimento nas áreas de fonoaudiologia e psicologia (AMANF, 2008, 25).
Contudo existem outros tipos de estudos para ajudar amenizar os incômodos sentidos pelos portadores ou até mesmo pela vaidade ou pra se sentir bem na sociedade que infelizmente é julgada pela aparência. A NF1 prejudica o desenvolvimento e deformação dos ossos causando o retardamento da puberdade, os tumores alguns precisam de várias sessões de cirurgia e mesmo assim não são totalmente definitivas mesmo fazendo a cirurgia pode ressurgir, podem causa problemas como: respiratórios, visão, coração, diversos órgãos, pois os tumores podem ser tanto por externos quanto internos do corpo. Além dos problemas cognitivos e as dificuldades psicológicas são frequente entre as pessoas com a NF1 são forçadas a encarar ao fato que possuem a doença (AMANF, 2008).
No dia 19 de novembro de 2013, um homem de 53 anos que possui a doença NF, sofreu uma desfiguração na fase por causa dos tumores, relata o incrível acontecimento em sua vida, nesse dia ele foi abraçado pelo papa Francisco e ele falou: - “Que se sentiu no paraíso”. O italiano que possui a doença genética, que sua mãe também possuía e veio a falecer, imaginou que o papa não iria abraça-lo porem mesmo sem saber se a doença e contagiosa ou não o papa não pensou duas vezes e abraço o homem com toda sua humildade. O italiano já passou por inúmeras cirurgias no coração, olhos e gargantas e tem muita dificuldade para respirar (TERRA, 2013).
As diversas características existentes da NF1, a presença de alterações
comportamental, têm sido constatadas por diferentes autores, além do déficit e as dificuldades de se relacionar com as outras pessoas, leva o portador a interpretar como herança da patologia, e principalmente quando a deformidade é visível (que pode causar discriminação social) seja por aparência ou lesão. O portador começa a se sentir isolado, completamente diferente da sociedade e a culpa e questiona o motivo de herdar essa patologia e consequentemente apresentam comportamentos de ansiedade, medo, baixo autoestima e que também afeta a vida profissional e principalmente a família. Quando criança tende a se isolar mais ainda dos amigos, pois falta informação, divulgação e quando adulto, passa por grandes transtornos psicossociais (JOHNSON., et al, 2005).
6. CONSIDERAÇÃO FINAL
A NF apresenta um caso clínico e complexo que requer uma equipe multidisciplinar para tratar portadores e seus familiares ao longo da vida. Grandes trabalhos têm sido realizados a mais de 10 anos para ajudar no tratamento, porém não existe ainda um tratamento eficaz para NF. Estudos moleculares são fatores primordiais para o diagnóstico precisos correlacionados a várias manifestações sistémicas apresentadas aos diferentes portadores, independente dos critérios estabelecidos por NIH.
Assim um dos métodos estudados seria o conhecimento da correlação entre fenótipo e genótipo, que ajudará no aconselhamento genético dos portadores e familiares em uma futura gestação. Por se tratar de uma patologia antiga e com várias características sistêmicas que ainda não tem cura e que acaba dificultando em alguns casos um tratamento adequado.
No Brasil possui várias Associações e uma delas citada no decorrer da bibliografia, na qual auxiliam portadores e seus familiares com NF por meio de múltiplos profissionais capacitados tais como, médicos e psicólogos, informando a conduta como cada família deverá proporcionar seja em escolar e na vida socioeconômico para obter um futuro promissor para os portadores com essa patologia.
7. REFERÊNCIA
AMANF. Lidando com a NF: um guia para adolescentes com neurofibromatose. Minas Gerais: Cartilha NF, 2008. 1-2 p.Disponível em
. Acessado em 05. mai. 2017.
BRUCE R, Korf, GRETCHEN, Shneider. Um guia para adolescentes com neurofibromatose. Minas Gerais: AMANF, 2008. 1-2 p.
BRUNO, Rebouças Tamassi. Avanços recentes em neurofibromatose. Neurinoma, São Paulo, v. 1, p. 1-8, set. 2003. Disponível em:
. Acessado em 20. abr. 2017.
CANALE Bebin et al. Von Recklinghausen disease of the nervous system. Anais Brasileiros de Dermatologia, Rio de Janeiro, v. 14, p. 132-162, 1972. Disponível em:< http://www.scielo.br/scielo.php?script=sci_nlinks&pid=S0365- 0596201300030032900004&lng=en>. Acesso em: 19 out. 2017.
CANCRO, na Família. Riscos em BRCA1. 2012. Portugal: Projeto de produção de informação do Programa Harvard Medical School, 2012.
DARRIGO, Luiz Guilherme Junior et al. Neurofibromatose tipo 1 na infância: revisão dos aspectos clínicos. São Paulo, v. 26, n. 2, p 176-182, Jun. 2008. Disponível em: . access on 19 Oct. 2017.
http://dx.doi.org/10.1590/S0103-05822008000200014>. Acesso em: 24 jun. 2014.
GUTMANN, Daivid et al. Neurofibromatosis Type 1: Modeling CNS Dysfunction. Washington, v. 41, n. 32, p 14087–14093, out. 2012. Diponível em:
. Acesso em: 19 set. 2017.
DeBELLA, et al. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Vancouver, v. 3, n. 105, p 608-614, mar. 2000. Disponível em: . Acesso 15 ago. 2016.
DEN DUNNEN, JT; Van OMMEN, GJ. The protein truncation test: A review.Leiden, v. 2, n. 14, p 95-102, jul. 1999. Disponível em:
10.f02t04>. Acesso 4 fer. 2016.
ACNefi. Consejo genético em las Neurofibromatosis: Los genes de la NF1 y de la NF2 – Disponível em: Acessado em: 07. mai. 2017.
FERNER RE; GUTMANN DH. Neurofibromatosis type 1 (NF1): diagnosis and management. London, n. 115, p. 939-955, Ago. 2013. Disponível em:
. Acesso em: 07.mai. 2017.
GUELLER Mauro. Neurofibromatose – Clínica, genética e terapêutica. 1 ed. São Paulo: Livro, 2004.
HANNA Hathaway NA et al. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. 2006, 127(1): 99-111.
DIAS IS; PESSOA SG; MACEDO JE; CAVALCANTE DJ et al. Abordagem
cirúrgica de neurofibroma gigante. 2012, 2(27): 336-339.
DR MCDONALD-MCGINN-DONNA - Dr ZACKAI ELAINE. Síndrome de deleción 22q11.2. 2017.dez.2012. Disponível em: < http://www.orpha.net/consor/cgi- bin/OC_Exp.php?Expert=567&lng=ES>. Acesso em: 07. mai. 2017.
KORF BR. Diagnostic outcome in children with multiple cafe au lait spots. 1992, 90(6): 924-927.
MAUTNER VF, KLUWE L, THAKKER SD, LEARK RA. Treatment of ADHD in
neurofibromatosis type 1. 2002, 44(3): 164-170.
MESSIAEN L, RICCARDI V, PELTONEN J, MAERTENS O et al. Independent NF1
mutations in two large families with spinal neurofibromatosis. 2003, 40(2): 122- 126.
NEUROFIBROMATOSE. Conference statement.National Institutes of Health Consensus Development Conference. 1988. 45(5): 575-578.
NIELSENGP, STEMMER-RACHAMIMOV AO, INO Y, MOLLER MB, ROSENBERG
AE, LOUIS DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. 1999. 155(6):1879-1884.
REV BRAS CLIN MED. Atualidades da neufibromatose tipo 1. Jun. 2008. Disponível em: . Acesso em: 25.nov. 2017.
RICCARDI VM. Neurofibromatosis: phenotype natural history and pathogenesis. 1992. 67(1): 264.
SMITH RW JR. The dermal oleastoses. Arch Dermatol. 1963, 3(88): 382-392.
SOUZA JF. Neurofibromatose tipo 1: mais comum e grave do que se imagina. 2009, 55(4): 394-399.
STUMPF DA; ALKSNE JF; ANNEGERS JF. Neurofibromatosis: conference statement. Arch Neurol. 1988, (45): 575-578.
XU GF,XU GF; O’CONNELL P; VISKOCHIL D; CAWTHON R et al.. The neurofibromatosis type 1 gene encodes a protein related to GAP. 1990, 62(3): 599–608.
JOHNSON H, WIGGS L, STRES G, HUSON SM: Psychological disturbance and sleep disorders in children with neurofibromatosis type 1. Development Medicine and Child Neurol 2005;47:237-242.
PERI CLAUDIO .dez-2015-UOL. Homem desfigurado abraçado pelo papa diz que “se sentiu no paraíso”. Dez.2015.Disponível em: . Acesso em: 17.set.2017.
Sabbagh A, Pasmant E, Imbard A et al: NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat 2013; 34: 1510–1518.
Por BARCELO Bruna; CORREIA Kamila. Aspectos clínicos, genéticos e psicossociais da neurofibromatose tipo 1.2017. 29 folhas. Trabalho de Conclusão de Curso de Biomedicina – Universidade de Cuiabá, Cuiabá, 2017.