RESUMO
É apresentada uma descrição da Síndrome de Li-Fraumeni (SLF), avaliando o impacto dessa síndrome na população do Paraná, dada sua alta prevalência nessa região, de 0,3%. Além disso, em 90% dos casos no Sul e Sudeste do Brasil, a SLF está associada mutação R337H no gene TP53. Por meio de revisões bibliográficas, foi possível identificar o efeito fundador na mutação R337H como sendo a causa da alta prevalência da síndrome no Paraná. O presente estudo também identificou a eficácia de triagens e vigilâncias para o aumento de qualidade de vida para os portadores da SLF. Por outro lado, dificuldades para esse cuidado também foram encontradas. A falta de estudos recentes sobre o assunto torna necessário o debate a respeito dessa síndrome tanto no meio acadêmico, quanto na sociedade em geral.
Palavras-Chave: Síndrome de Li-Fraumeni, TP53, mutação R337H
ABSTRACT
This work aims to describe the Li-Fraumeni Syndrome (LFS), assessing its impact on the population of Parana state, given the high prevalence in this region, of 0.3%. Moreover, in 90% of the cases in southern and southeastern Brazil, the LFS is associated with R337H mutation in the TP53 gene. Bibliographic reviews allowed the identification of a founder effect of the mutation R337H as the cause of that high prevalence of the syndrome in Parana state. This research also identified the effectiveness of screening and surveillance for the LFS carriers’ better quality of life. On the other hand, difficulties for that caring have been found. The lack of studies regarding this subject in recent years shows the need for this debate in academic field and in society.
Keywords: Li-Fraumeni Syndrome, TP53, R337H mutation.
Sumário
1. INTRODUÇÃO
1.1. Contexto do problema
O câncer é uma doença bastante estudada atualmente; porém, ainda existe um vasto campo para ser pesquisado, para que síndromes como a Li-Fraumeni, a qual acomete algumas pessoas ao redor do mundo, possa ser melhor compreendida. Esta é uma síndrome que, no Brasil, mais especificamente nos estados do sul e sudeste, está associada a mutação a R337H no gene TP53 encontrada com uma alta prevalência nos estados do Sul e do Sudeste em crianças portadoras daSLF que está associada com um aumento no risco de câncer(CUSTÓDIO et al, 2013).
Por conta disso, esta revisão bibliográfica tem como objetivo compreender melhor a Síndrome de Li-Fraumeni. Será avaliado como a mutação R337H tem sua frequência significativamente aumentada quando comparada com outros estados não-pertencentes às regiões sul e sudeste, levando em conta o provável efeito fundador.
Para levar este trabalho a bom termo, ele foi dividido em cinco partes. A primeira é a introdução, iniciada por esta contextualização do problema, e apresentará, a seguir, os objetivos e a justificativa do trabalho. Na segunda parte, descreve-se a metodologia utilizada nesta pesquisa. A terceira parte, que contém o referencial teórico, foi dividida em cinco subtópicos. O primeiro trata da Síndrome de Li-Fraumeni (SLF), explicando suas características e estudos encontrados a respeito da síndrome. Em seguida, no segundo subtópico, elaborou-se uma explanação do gene afetado pela SLF, o TP53, descrevendo-o em seu funcionamento normal e o que ocorre quando ele está mutado. O subtópico de número três apresentará a proteína p53, codificada pelo gene TP53. No subtópico seguinte, descreve-se as principais mutações no gene em questão, focando, especialmente, a mutação R337H. A razão desse foco será descrita no decorrer deste estudo. O quinto subtópico da terceira parte revisa estudos a respeito da SLF no estado do Paraná. No subtópico de número 6, trata-se das pesquisas que evidenciam a influência do efeito fundador na região. O último subtópico dedica-se a demonstrar a importância de triagens, acompanhamentos e diagnósticos para o tratamento e cura da síndrome.
A quarta parte consiste em conclusões e recomendações oriundas desta pesquisa. Por fim, a última parte, são as referências bibliográficas. A seguir apresenta-se os objetivos deste trabalho.
2. Objetivos
Avaliar o impacto da Síndrome de Li-Fraumeni na população do Paraná, considerando a alta prevalência da mutação R337H no gene TP53 e outras possíveis causas genéticas como mutaçõesnos éxons 5-8 do gene TP53.
2.1. Objetivos específicos
Para cumprir o objetivo deste trabalho, os seguintes objetivos específicos serão perseguidos:
-
Descrever a SLF e LFL;
-
Evidenciar o efeito fundador;
-
Revisar os casos no Paraná;
3. Justificativa
A síndrome de Li-Fraumeni, com alta prevalência no sul e sudeste do país, especialmente no Paraná, está associada ao maior risco de desenvolvimento de alguns tipos de câncer. Pesquisadores como Custódio, et al. (2013) afirmam que para um diagnóstico precoce e maior eficácia no prognóstico são necessárias triagem, vigilância e heredograma. Portanto, esta revisão visa compreender a síndrome e sua alta prevalência no estado do Paraná, além de observar como a triagem precoce e o acompanhamento afetam o prognóstico.
4. Metodologia
O trabalho será do tipo exploratório, desenvolvido através de uma pesquisa bibliográfica em livros, teses e dissertações, além de artigos científicos, para descrever a Síndrome de Li-Fraumeni e seu impacto na população do Paraná.
5. REFERENCIAL TEÓRICO
5.1. Síndrome de Li-Fraumeni
A síndrome foi descoberta no ano de 1969 por dois médicos americanos, Frederick Pei Li e Joseph Fraumeni Junior, com base em um estudo que revisou 280 prontuários e 418 atestados de óbito de crianças norte americanas que tinham diagnóstico de rabdomiossarcoma, tumor que se desenvolve nos músculos, tendões e tecidos conjuntivos (Varley, 1997a). Buscando o histórico familiar dos pacientes, verificaram que dentre as famílias estudadas, 5 apresentaram uma alta prevalência de tumores nas gerações que foram avaliadas, incluindo diagnóstico de sarcoma e casos de câncer de mama em idade jovem (LI e FRAUMENI, 1969a). As famílias também relataram a presença de outros tipos de tumores em idades mais avançadas. A partir das análises do agrupamento pouco comum de tumores malignos em idade jovem, percebeu-se um padrão de transmissão hereditária. Com base nesses estudos, os autores Li e Fraumeni propuseram uma nova síndrome de câncer familiar relacionada a diversos tipos de tumores: a Síndrome de Li-Fraumeni (ACHATZ, 2008).
A Síndrome de Li-Fraumeni (SLF, OMIM #151623) é uma predisposição hereditária ao câncer de caráter autossômico dominante; isto é, não há diferenças entre a herança entre homens e mulheres, e basta que o indivíduo herde um alelo mutado para que esteja predisposto a manifestar os sintomas da doença (VARLEY et al., 1997b). Essa síndrome é caracterizada clinicamente pelo risco aumentado no desenvolvimento de diversos tipos de câncer, frequentemente em idade jovem, tais como câncer de mama pré-menopausa, sarcomas de partes moles e osteosarcomas, leucemias, tumores do sistema nervoso central e adrenocorticais, câncer de pulmão, pâncreas, entre outros (GUHA e MALKIN, 2017; ACHATZ, 2008). A SLF também é tida como uma síndrome de alta penetrância, cujo risco cumulativo de câncer é de 50% aos 40 anos de idade e de até 90% aos 60 anos (ACHATZ, 2008).
O diagnóstico clínico da síndrome de Li-Fraumeni (SLF) é definido pela presença de um indivíduo diagnosticado com sarcoma antes dos 45 anos de idade, um parente de primeiro grau com câncer antes dos 45 anos de idade e um segundo parente, este de primeiro ou segundo grau, diagnosticado com câncer antes dos 45 anos de idade ou sarcoma em qualquer idade (LI et al., 1988). Também foram observadas famílias que apresentavam algumas características da síndrome, mas não a expressão do fenótipo completo. Essas famílias passaram a ser denominadas como portadoras da Síndrome de Li-Fraumeni like (LFL), a qual é definida pelo probando com um tumor do espectro LFS antes dos 45 anos, e mais um parente de primeiro ou segundo grau que também tenha apresentado algum câncer do espectro SLF antes dos 60 anos (BIRCH et al., 1994). Há também uma definição alternativa de LFL, ainda menos restrita, que considera a presença de dois tumores diferentes relacionados ao SLF em parentes de primeiro ou segundo grau, mas em qualquer idade (EELES, 1995). A idade média de câncer na SLF é mais tardia que na LFL (ROSSI et al., 2015).
A Síndrome de Li-fraumeni, está associada a mutações no gene supressor de tumor que codifica para a proteína p53 (TP53) em células germinativas (FETT-CONTE; SALLES, 2002). Portanto, é importante conhecer o gene e na próxima seção falaremos sobre ele.
5.2. O gene TP53
No final da década de 80, Finlay et al. (1989), identificaram o gene TP53 como um supressor de tumor. Esse gene, que apresenta 11 éxons, está localizado no braço curto do cromossomo 17 (17p13.1), figura 01 (fig. 01), e codifica uma proteína nuclear tetramérica de 53 kDa (quilodaltons) que atua como fator transcricional (FETT-CONTE e SALLES, 2002; ACHATZ, 2008; CORREA, 2016), produzindo um mRNA de aproximadamente 2500 pb (ACHATZ, 2008).
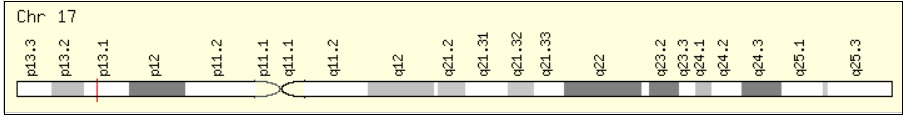
Fonte: https://www.genecards.org/cgi-bin/carddisp.pl?gene=TP53&keywords=p53
Conhecido como “guardião do genoma”, o gene TP53 desenvolve uma função central na manutenção da integridade do genoma, sendo que sua expressão está envolvida com o controle da proliferação e da morte celular, em resposta a vários sinais, incluindo danos ao DNA. As situações de estresse que podem ativar a transcrição do TP53 são, por exemplo, radiação ionizante ou ultravioleta, hipóxia, encurtamento telomérico, dano ao DNA e sinalização oncogênica (Kastan et al., 1991; 2003; Shulin Harris e Levine, 2005; 2003; Oren, 2003; Levine e Oren, 2009). Após a tradução do mRNA, a proteína tem um tempo de meia vida curto, durando cerca de 10 a 30 minutos, e, em condições de homeostase celular, seus níveis são mantidos baixos (Picksley e Lane, 1993; Kubbutat et al., 1997).
A perda de função do gene TP53, que pode ocorrer por mutações, está associada ao câncer em humanos. Mais de 75% das mutações em TP53 conduzem à expressão de uma proteína de comprimento completo com uma única substituição de aminoácido (BROWN et al., 2009).
5.3. A Proteína p53
A p53 é uma fosfoproteína nuclear que desempenha um papel fundamental na regulação do ciclo celular, particularmente na inibição da proliferação celular. Trata-se de uma proteína composta por um homotetrâmero, cujas subunidades possuem 393 aminoácidos cada. Divide-se cada monômero em cinco domínios bem definidos (I-V), os quais têm uma função importante na regulação da proteína (fig. 02). O primeiro é o domínio amino-terminal (N) de transativação, o segundo rico em prolina, o terceiro de ligação ao DNA, o quarto domínio é de oligomerização e o quinto de regulação carboxi-terminal (C) (ACHATZ, 2008; LÓPEZ et al, 2006).
Segundo López et al. (2006), o domínio I corresponde aos domínios de ativação 1 e 2 (Activation Domain 1 and 2 - AD1 e AD2), responsáveis pela ativação da proteína. O domínio II, rico em prolina (Proline Rich Domain - PRD), está envolvido na atividade supressora da p53 e é responsável pela ativação da proteína; observa-se que também está envolvido no processo de apoptose da célula. O domínio III está envolvido no reconhecimento e na ligação à sequência específica no DNA (DNA Binding Domain - DBD). Esse domínio de ligação contém pontos quentes de mutação (hot spots); muito importantes, pois mutações nessa região podem comprometer a capacidade da proteína p53 se ligar a sequências-alvo no DNA. Os códons 248 e 273 são os códons de contato com o DNA, e os códons 175, 249 e 282 são os que dão estabilidade à proteína. Os sítios citados acima estão envolvidos com 25% dos cânceres que apresentam a mutação da p53. O domínio IV (Tetramerization Domain - TD) é responsável pela tetramerização das subunidades da p53; essa é uma região de alfa-hélice, e as moléculas da p53 com mutações neste local tornam-se mais instáveis, afetando a ligação ao DNA. O V e último domínio é o alcalino (DB), necessário para regulação da atividade da p53. Os domínios estão demonstrados na figura 02.
Figura 2: DOMÍNIOS DE I-V DA PROTEÍNA p53.

Fonte: Adaptado de López; Sánchez; Salcedo (2006).
A via da p53 é composta por uma rede complexa de proteínas que evoluíram para responder a diferentes sinais de estresse, principalmente a danos ao DNA. As funções principais da proteína envolvem sua atuação como fator de transcrição, bem como a interação com outras proteínas a partir da formação de complexos (Kern et al., 1991; Farmer et al., 1992; Zhao et al., 2000).
A expressão dos genes regulados pela p53 e a atuação dessa proteína em complexos depende da natureza e da intensidade do estresse, do tipo celular e do microambiente em que a célula está inserida. Assim, a proteína pode regular diferentes processos celulares de acordo com a situação específica em que a célula se encontra. Os mecanismos controlados pela p53 são, por exemplo, proliferação, senescência, parada do ciclo celular, reparo de dano no DNA, apoptose e autofagia (FETT-CONTE e SALLE, 2002).
A proteína suspende o ciclo celular de células que apresentam danos ao DNA, a fim de corrigir o erro e então liberar novamente a célula para que ela continue sua divisão normalmente após a correção (FETT-CONTE e SALLE, 2002). Em outra maneira de ação para interromper o ciclo celular em resposta a algum dano celular, a p53 atua conjuntamente com outros seis genes, ligando-se ao promotor do gene p21, do qual o produto proteico é um inibidor de quinase dependente de ciclina, que inibe a proteína Retinoblastoma (pRb) pela quinase dependente de ciclina 4 (CDK4). Essa interação promove a parada do ciclo celular na fase G1, antes da fase S onde ocorre a duplicação do DNA (FETT-CONT e SALLE, 2002). Uma outra forma da proteína p53 atuar é fazer check point (ponto de checagem) de S para G2, dependente do domínio C-terminal (FETT-CONTE e SALLE, 2002). Uma desregulação do crescimento celular nas fases G1 e G2, ocasionada por um erro de checagem da p53, pode contribuir para o desenvolvimento de muitos tipos de cânceres. Tais células, que perderam essa característica de regulação do crescimento, são denominadas transformadas (SHERR, 1996).
A parada do ciclo celular na fase G1, antes de ocorrer a duplicação do DNA (fase S), permite o reparo do DNA danificado. Uma alternativa de atuação da p53 a danos não reparados, caso a via com a proteína pRb não seja reparada totalmente, é aquela na qual ocorre a indução para a morte celular programada (apoptose) pelas proteínas sensoras de checagem (FETT-CONTE e SALLE, 2002).
A proteína p53 tem papel essencial na regulação do ciclo celular em resposta a um dano, como descrito acima. Quando ocorre uma mutação no TP53, a proteína pode perder seu papel de correção em um evento de estresse celular, este sendo tanto externo quanto interno (GIACCIA e KASTAN, 1998).
5.3.1. Mutações do Gene TP53
Segundo Weinberg (2008), foram realizados estudos em 1987 que demonstraram que alelos do TP53 com mutações pontuais são comuns no genoma de uma ampla variedade de células de tumores em humanos. Pesquisas com dados até o ano de 2002 demonstraram que o gene TP53 está mutado em 30 a 50% dos cânceres humanos que ocorrem com mais frequência (WEINBERG, 2008).
Mutações no gene TP53 geram variabilidade somática na sequência codificante, que estão relacionadas com 33% de todos os tumores malignos esporádicos, podendo chegar até 50% de todos os tumores invasivos (GIACOMAZZI, 2012). Cerca de 84% das mutações vistas no gene TP53 ocorrem no domínio de ligação ao DNA, codificado pelos éxons 5 a 8 do gene TP53, (GIACOMAZZI, 2012). Segundo Giacomazzi (2012), no banco de dados R15 do Internatonal Agency for Research on Cancer, 2012 (IARC) foram descritas 597 mutações germinativas, 27.580 mutações somáticas e 85 polimorfismos no TP53.
Segundo Weinberg (2008), as células tumorais que são mutantes no loco do TP53 podem sofrer perda de heterozigosidade, dessa forma o alelo tipo selvagem é descartado, produzindo uma célula com dois alelos TP53 mutantes.
Normalmente a proteína p53 atua como um fator transcricional homotetramérico. Porém, uma célula que carregue um único alelo mutado, portanto heterozigota para o gene TP53, vai expressar os dois alelos – o selvagem e o mutado. Quando as quatros subunidades da proteína forem montadas, a cadeia polipeptídica de cada subunidade será selecionada ao acaso. Assim há uma chance de apenas 1/16 de todas as combinações possíveis serem de fato funcionais, ou seja, de as quatro subunidades virem do alelo selvagem (Weinberg, 2008). As demais 15/16 combinações possíveis levarão ao menos uma subunidade mutada, o que pode reduzir a estabilidade e/ou funcionalidade da proteína.
O domínio de ligação ao DNA da p53, contido nos éxons 5-8 (fig. 02), corresponde à região mais estudada e relacionada com a SLF/LFL, sendo que a maioria das mutações patogênicas nessa região são mutações de ponto que alteram a sequência específica de ligação (YAN e CHEN, 2010). Entretanto, foram identificadas mutações no gene TP53 fora da região de ligação ao DNA, assim como rearranjos e deleções que expandiram as possibilidades de mutações neste gene (OLIVIER et al., 2010; KAMIHARA et al., 2014).
A prevalência das SLF e LFL ainda não foi estabelecida no mundo. Porém, a partir de estudos conduzidos nos Estados Unidos e na Europa, foi estimado que as mutações germinativas no gene TP53 ocorrem em uma frequência aproximada de 1 em 5.000 indivíduos (LALLOO et al., 2003; GARBER e OFFIT, 2005). Já no Brasil, há uma prevalência relativamente alta da síndrome devido à presença de uma mutação fundadora, que causa uma substituição do aminoácido Arginina (Arg) por uma Histidina (His) na posição 337 da proteína p53, a qual denomina-se R337H. Essa mutação parece ocorrer em 0,3% da população do Sul do Brasil (ACHATZ, 2008). Dada essa alta prevalência e o objetivo deste trabalho, de avaliar o impacto da SLF na população do Paraná, ressaltar-se-á a mutação R337H na seção a seguir.
5.3.2. Mutação R337H TP53
Em 1998, a mutação R337H foi descrita pela primeira vez em uma família na França (ACHATZ, 2008) sendo que três anos depois, em 2001, ela foi identificada no Brasil por Ribeiro et al. (2001), ao analisarem 36 pacientes com carcinoma pediátrico adrenocortical tratados no Paraná, dos quais 35 apresentavam a mesma mutação germinativa R337H.
A mutação R337H não está localizada no domínio de ligação ao DNA do gene TP53, como a maioria das mutações conhecidas no TP53 relacionadas às síndromes de Li-Fraumeni e Li-Fraumeni like. Essa mutação, que ocorre no códon 337, no éxon 10 (fig. 02) do TP53, corresponde a uma região que participa da oligomerização da p53 e leva a uma mudança de arginina para histidina no local citado (R337H – CGC para CAC no códon 337) (ACHATZ, 2008; CUSTÓDIO et al., 2013). Essa mutação foi relacionada com a maior incidência de tumores adrenocorticais (ACTs) encontrado em crianças no Brasil. A maioria dos portadores eram menores de 15 anos, sendo que em idade avançada o mais comum eram tumores de pulmão, cérebro e estômago. Os ACTs são mais agressivos quando R337H está presente em homozigose em crianças (CUSTÓDIO et al., 2013). A principal consequência desta característica é que, ao contrário da esperada seleção negativa durante a evolução, seus portadores possuem maior probabilidade de viver até idades reprodutivas, possibilitando a propagação desta mutação e sua permanência ao longo de gerações (GARRITANO et al., 2009).
Garritano et al. (2009) concluíram que seu estudo deve ser visto com parcimônia, pois os critérios que foram utilizados para avaliações familiares são muito amplos. Nas famílias que preencheram os critérios da SLF, o risco que está associado a R337H parece ser o mesmo encontrado em outras mutações.
DiGiammarino e colaboradores (2002), a partir de uma análise cromatográfica in vitro da estrutura e estabilidade da p53-R337H, viram que elas são muito semelhantes à proteína p53-wt (proteína 53 wild type). No entanto a p53-R337H dependia da estabilidade do pH da célula, sendo a molécula menos estável em pH alcalino. Em temperatura de 37°C e pH 8, cerca de 70% da p53-R337H encontrava-se desnaturado, no entanto a p53-wt desnaturava apenas a 50ºC, independente do pH a qual fosse submetida. Foi observado posteriormente que na proteína selvagem se formava uma ponte de sal, formada por uma ligação de moléculas com cargas opostas, entre os aminoácidos Arginina 337 (Arg 337), de carga positiva, e Asparagina 352 (Asp352), negativa. A ponte é essencial para dimerização e posteriormente a formação do tetrâmero. Já na proteína mutada, a Arginina (R) é substituída por uma Histidina (H) na posição 337, induzindo diferentes mudanças químicas nessa região. Assim, há uma desestabilização nas pontes salinas, desestruturando o tetrâmero conforme demonstra a fig.03 (GIACOMAZZI, 2012).
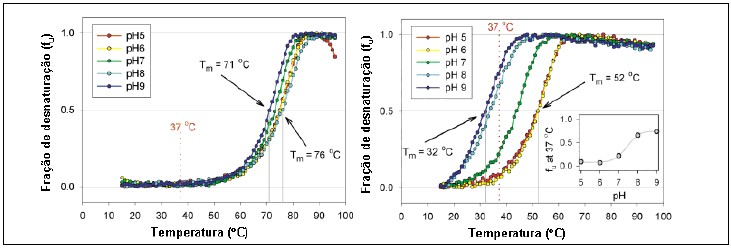
Fonte: Giacomazzi (2012).
Portanto, a desestabilização da oligomerização da proteína irá afetar indireta e negativamente a ligação de p53 aos domínios de ligação no DNA dos genes-alvo, afetando sua função. Dito isso, analisar-se-á, no item a seguir, a alta prevalência dessa mutação no estado do Paraná.
5.4. Revisão dos casos no estado do Paraná (PR)
As SLF e LFL na população do estado do PR tem uma alta prevalência da mutação R337H TP53 associadas a tumores adrenocorticais (ACTs) pediátricos (CUSTÓDIO et al., 2013). A mutação R337H no gene TP53, está associada no Sul e Sudeste do Brasil de 90% a 97% das mutações encontradas nesse gene. No Paraná aproximadamente, 27% das famílias com outros tumores (principalmente de mama e gastrintestinais) preencheram, nos estudos, parcialmente os critérios para SLF ou seja, elas são LFL (DUDEQUE PIANOVSKI e FIGUEIREDO, 2017).
Foram realizados estudos por Custódio et al. (2013) sobre o impacto da triagem neonatal e vigilância da mutação TP53 R337H no estado do PR. Nesse estudo, foram testados 171.649 recém-nascidos, sendo que 461 (0,27%) carregavam a mutação R337H, demonstrando uma prevalência dessa mutação para a população estudada.
Os resultados obtidos por Custódio et al. (2013) demonstraram que, das crianças heterozigotas para a mutação (461), 11 desenvolveram ACTs. Portanto, a penetrância média estimada foi de 2,39%. Cumulativamente, a penetrância média aos 2 anos de idade foi de 0,7%; aos 5 anos, de 2,21%. Esses dados corroboram com o fato de que os ACTs associados a R337H desenvolvem-se durante os primeiros 5 anos de vida.
Os autores concluíram que a triagem e a vigilância foram altamente efetivas para a detecção de ACTs, uma vez que o valor preditivo negativo foi de aproximadamente 100% (apenas 2 casos dos 171.188 recém-nascidos triados como negativos para a R337H desenvolveram ACTs). Além disso, a abordagem utilizada pelos autores, com base na predisposição genética a ACTs, permitiu um monitoramento sensível nos primeiros anos do tumor (CUSTÓDIO et al. 2013).
Os resultados sugerem que de 90 a 95% das crianças portadoras de ACTs poderiam ser curadas como resultado de triagem e acompanhamento, sem os quais apenas 50% das crianças com ACTs sobrevive e muitos requerem uma quimioterapia intensiva e tóxica (CUSTÓDIO et al. 2013).
O estudo de Mathias (2014) analisou as frequências da mutação R337H em TP53 em casos de carcinoma mamário, atendidas no Hospital Nossa Senhora das Graças de Curitiba, PR. Embora as mutações nos genes Breast Cancer 1 (BRCA1), BRCA2, TP53, Fosfatase e homólogo de tensina no cromossomo 10 (PTEN) e CheckPoint Kinase 2 (CHEK2) sejam as principais causadoras de câncer mamário esporádico, as Síndromes de Câncer de Mama e Ovário Hereditário (HBOC) e a SLF são as que caracterizam o câncer de mama hereditário. No trabalho, a autora observou a presença ou ausência da mutação R337H no gene TP53 em amostras de pessoas do sexo feminino com diagnóstico de câncer de mama. Foram usados vários métodos de análises moleculares nas amostras obtidas. Foram genotipadas 314 mulheres com câncer de mama, dessas, uma (0,32%) era portadora da mutação R337H no gene TP53, demostrando que a frequência era a mesma da população em geral.
Piovezan (2006) concluiu que existe uma alta prevalência da mutação R337H no gene TP53 no estado do PR, com uma prevalência uniforme ao redor de 2,2 a 2,5/1000. O autor observou que não houve mutação “de novo” – uma mutação que não foi herdada por nenhum dos pais – nos recém-nascidos avaliados. O pesquisador também verificou que a distribuição do alelo TP53 R337H nos grupos étnicos mostra uma predominância em indivíduos de pele branca, sendo que não foi observado o alelo em pessoas japonesas, chinesas ou de outra etnia oriental. Outras pesquisas (PINTO et al., 2004; GARRITANO et al., 2009; OLAZAR, 2013; CHAMPRET et al, 2000; RIDLEY, 2006) puderam inferir o efeito fundador como causa dessa predominância. A subseção a seguir apresenta esses trabalhos que demonstram o efeito fundador.
5.5. Efeito fundador
Pinto et al. (2004), sugeriram que a mutação R337H foi estabelecida na população brasileira a partir de um ancestral comum, defendendo a hipótese da ocorrência do efeito fundador, a partir da análise de dois marcadores polimórficos intragênicos, em pacientes com tumores adrenocorticais que eram portadores da mutação R337H em TP53.
Em estudos subsequentes no Sul do Brasil, Garritano et al. (2009) puderam observar que desde a descoberta inicial da alta incidência da mutação no gene TP53 pR337H em crianças com ACTs no Brasil, há muitas controvérsias da origem da mutação e o impacto da predisposição ao câncer familiar. No artigo foi analisado a estrutura do haplótipo do locus do TP53 para identificar precisamente os haplótipos que carregam a mutação R337H e, através das análises, puderam sustentar a hipótese do efeito fundador.
Para isso, Garritano et al. (2009) utilizaram resequenciamento total no locus do TP53 e genotipagem de polimorfismos de nucleotídeos únicos (SNPs) de alta densidade para refinar as definições de haplótipos do locus do TP53. Os polimorfismos de nucleotídeo único ou SNPs é uma forma de variação (0,1%) no genoma humano, isso ocorre quando há substituição de uma única base adenina, citosina, guanina e timina (A,C,G e T) em um determinado locus cromossômico (OLAZAR, 2013). Foram analisadas 47 amostras de DNA. Seis amostras foram extraídas de células cancerosas cujas linhagens tinham sido previamente estabelecidas em laboratório, tipadas como hemizigotas para TP53 com subsequente perda de heterozigosidade. Outras 15 amostras de DNA eram provenientes de células linfoblastóides cancerosas extraídas de pacientes com câncer de mama; 25 eram de pacientes brasileiros com SLF, coletadas na região de São Paulo, e uma de Chimpanzé. Para todas as amostras humanas, os doadores eram caucasianos, entretanto a presença de cromossomos não-caucasianos não pode ser excluída das amostras brasileiras (GARRITANO et al., 2009). A análise de haplótipos de 12 portadores da R337H não relacionados, usando um conjunto de 29 marcadores de sequência genética (tags) de SNPs, revelou que todas as pessoas tinham o mesmo haplótipo, sendo que a presença da mutação nesse haplótipo foi confirmada por PCR alelo específica. Como as análises tinham a mesma sequência de SNPs, há uma forte sugestão de efeito fundador. A análise dos padrões de 103 tumores diagnosticados em 12 famílias estudadas demonstrou que a presença de R337H está associada com múltiplos cânceres do espectro da SLF, com baixa penetrância antes dos 30 anos. As famílias com R337H estão bem distribuídas ao longo do eixo historicamente conhecido como a rota principal usada por mercadores portugueses originários no século XVIII e XIX. A circunstância histórica e a baixa penetrância antes dos 30 anos pode ter contribuído para que crianças cheguem a idade adulta e consigam passar seus genes para seus descendentes (GARRITANO et al., 2009).
Após a utilização de alguns métodos e análise, eles puderam observar que a hipótese mais provável é que a R337H surgiu em um indivíduo de descendência europeia. O que suporta este cenário é que apenas um caso foi encontrado fora do Brasil, diagnosticado na França, em uma menina de 8 anos com ACT e com ancestralidade portuguesa (CHAMPRET et al., 2000).
No trabalho de Garritano et al. (2009), o resequenciamento nos locus do gene TP53 permitiu investigar mais SNPs informativos e uma maior resolução no mapa de haplótipos do que estudos anteriores. Com isso, o alelo R337H acaba por ser um representante único de uma mutação patogênica fundadora, a qual persiste em uma população grande e heterogênea. Possívelmente, a persistência seja explicada pelos cenários históricos específicos da incorporação do alelo e sua penetração moderada de início precoce, em vez de por segregação cultural ou geográfica na sua população de acolhimento (GARRITANO et al., 2009).
Segundo GARRITANO et al. (2009), a maioria das pessoas diagnosticadas com SLF/LFL no Brasil descende de imigrantes portugueses e moram nos estados do Rio de Janeiro, São Paulo, Paraná, Santa Catarina e Rio Grande do Sul. Porém, como há décadas as pessoas desses estados migram para outras regiões, os moradores de outras regiões podem ter essa síndrome e nunca terem sido diagnosticados. No Brasil, há uma prevalência relativamente alta da síndrome devido à presença de uma mutação fundadora (GARRITANO et al., 2009). As mutações fundadoras estão divididas em 2 segmentos. O primeiro é o estabelecimento de uma população nova por um grupo pequeno de fundadores, sendo assim, chamado de efeito fundador. No segundo segmento, os fundadores são uma porção limitada da variação genética. Para ser um efeito fundador completo é necessário que tenha fundadores que sejam geneticamente não representantes da população de origem (RIDLEY, 2006). Para melhor compreender as consequências do efeito fundador, a realização de triagens, acompanhamentos e diagnósticos torna-se necessária. Além disso, tais ferramentas permitem uma melhora na qualidade de vida dos portadores.
5.6. Triagem, acompanhamento e diagnóstico
Para Piovezan (2006), realizar mapeamentos, estudos moleculares e epidemiológicos podem esclarecer melhor a penetrância da mutação R337H em casos de ACT e outros tipos de câncer no PR. Conhecendo os indivíduos portadores da mutação R337H, seria possível dar um acompanhamento mais específico e com frequência para os recém-nascidos e adultos, podendo se oferecer um tratamento precoce e curativo do ACT. No projeto, ficou em aberto a investigação sobre a participação ambiental na origem da mutação, embora não tenha ocorrido a mutação “de novo” quando o estudo foi realizado.
O trabalho de Pianovski e Figueiredo (2017), que estudou 600 famílias no estado do PR, detectou a SLF em menos de 5% delas. Foi surpresa para os autores a ausência de câncer em várias dessas famílias. Esse dissenso demonstra que o aconselhamento genético e o processo educativo devem se unir ao histórico de câncer, e ao heredograma completo de todos os testados para mutação em quatro ou mais gerações, sem pressupor de que a mutação R337H no gene TP53 por si só indica risco de câncer. A vigilância voltada para o diagnóstico precoce do ACT em crianças diagnosticadas com a mutação R337H no gene TP53, identificada na triagem neonatal, com utilização de hormônios pediátricos e ecografia abdominal, foi considerada eficiente, mas também o treinamento educativo com os pais, focado nas manifestações clínicas, se mostrou eficaz.
Os avanços das pesquisas e tecnologias prometem mudanças no diagnóstico, tratamento e aconselhamento genético, a fim de que medidas sejam tomadas antes mesmo da definição do tratamento, permitindo assim métodos menos invasivos, uma melhor qualidade de vida e uma maior sobrevida para as pessoas acometidas pelo câncer (KAVALEC, 2006).
Apesar dos bons resultados advindos da vigilância, existem várias dificuldades a serem enfrentadas. Custódio et al. (2013) identificaram alguns desafios como nível sócio econômico, baixa educação, longas distâncias dos centros de estudo, impossibilidade de deixar o trabalho e a percepção do ACT ter uma penetrância baixa.
6. Conclusões e recomendações
Fazendo essa pesquisa bibliográfica percebeu-se que devido a alta prevalência da síndrome no PR, será necessário continuar com mais estudos para aprimorar os conhecimentos e diagnósticos. Além disso, com bases nos trabalhos apresentados por esta pesquisa, foi possível identificar a eficácia de vigilância e triagem contínuos no aumento da qualidade de vida para os portadores da síndrome.
Foi observado, ainda, a dificuldade que as pessoas têm de acesso ao tratamento por morarem em locais de difícil acesso, por exemplo, como identificado por Custodio et al. (2013). Outro fator muito importante é a falta de percepção do problema e sua gravidade. Por ser uma síndrome de penetrância baixa e muitos portadores não manifestarem sinais clínicos cedo, já que são assintomáticos, indivíduos acometidos pela SLF chegam à idade fértil e passam seus genes para prole. Dessa maneira, os portadores de mutações no gene TP53 podem não levar em conta, na opção de paternidade ou maternidade, a chance de transmitir a mutação do TP53 a seus filhos.
Traçar heredogramas para analisar a ancestralidade da síndrome possui também uma significância alta. Essa metodologia permite saber os casos de cânceres na família e orientar alguns cuidados diários como alimentação mais saudável, não se expor a radiações como ultravioleta, raios-X, entre outros, permitindo, assim, que a pessoa possa ter uma expectativa de sobrevida maior com mais qualidade, ou até mesmo nem venha desenvolver o câncer.
7. REFERÊNCIAS BIBLIOGRÁFICAS
ACHATZ, M.I.A.S.Z. Diagnóstico Molecular da Síndrome de Li-Fraumeni em Famílias Brasileiras. Dissertação de Mestrado. Fundação Antônio Prudente. São Paulo, 2006.
ACHATZ, M.I.A.S.Z. Modificadores de penetrância de mutações germinativas no gene TP53 em famílias brasileiras com diagnóstico clínico da síndrome de Li-Fraumeni e Li-Fraumeni like: impacto dos polimorfismos intragênicos do TP53 e de genes que regulam a atividade da p53. Tese de Doutorado. Universidade de São Paulo, 250pg, 2008.
BIRCH, J.M.; HARTLEY, A.L.; MARSDEN, H.B.; HARRIS, M.; SWINDELL, R. Excess risk of breast cancer in the mothers of children with soft tissue sarcomas. British Journal of Cancer, v. 49, p. 325-331, 1984.
BROWN CJ, LAIN S, VERMA CS, FERSHT AR, LANE DP. Awakening guardian angels: drugging the p53 pathway. Nature reviews Cancer, v. 9, p. 862-73, 2009.
CORREA, H. Li-Fraumeni Syndrome. Journal of Pediatric Genetics, v.5, n.2, p. 84-88, 2016.
CUSTODIO, G.; PARISE, G.A.; FILHO, N.K. et al. Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. Journal of Clinical Oncology, v. 31, n. 20, p. 2619, 2013.
DIGIAMMARINO, E.L; LEE A.S; CADWELL, C. et al. A Novel Mechanism of Tumorigenesis Involving PH - Dependent Destabilization of a Mutant p53 Tetramer. Nature Structural and Molecular Biology, v. 9, n. 1, p. 12, 2002
DUDEQUE PIANOVSKI, A.; DE FIGUEIREDO, B. C. Mutação R337H no gene TP53, frequente no Sul e Sudeste do Brasil, não parece ser suficiente como causa de câncer em crianças e adultos. Revista Brasileira de Cancerologia, v. 63, n. 3, p. 219-221, 2017.
EELES, R.A. Germline mutations in the TP53 gene. Cancer Surveys. V. 25, p.
101-124, 1995.
FETT-CONTE, A.C.; SALLES, A.B.C.F. A importância do gene p53 na carcinogênese humana. Revista Brasileira de Hematologia e Hemoterapia, v. 24, n.2, p.85-89, 2002.
FINLAY, C.A.; HINDS, P.W.; LEVINE, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell, v.57, n.7, p. 1083-1093, 1989.
GARBER, J.E.; OFFIT K. Hereditary cancer predisposition syndromes. Journal of Clinical Oncology, v. 23, n. 2, p. 276-292, 2005.
GARRITANO, S; GEMIGNANI, F; PALMERO, E. I et al. Detailed Haplotype Analyis at the TP53 Locus in pR337H Mutation Carriers in the Population of Southern Brazil: Evidence for a Founder Effect. Human mutation, v. 31, n. 2, p. 143-150, 2009.
GENETICS HOME REFERENCE. TP53 gene. Acesso em: 1 novembro, 2018 <https://ghr.nlm.nih.gov/gene/TP53>
GIACCIA, A.J.; KASTAN, M.B. The complexity of p53 modulation: emerging patterns from divergent signals. Genes & development, v. 12, n. 19, p. 2973-2983, 1998.
GIACOMAZZI, J. Prevalência da Mutação Germinativa TP53 p.R337H em Indivíduos com Tumores no Espectro da Síndrome de Li-Fraumeni. Dissertação ao Programa de Pós-graduação para Medicina, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre (RS), p. 65 -114.
GUHA, T.; MALKIN, D. Inherited TP53 Mutations and the Li-Fraumeni Syndrome. Cold Spring Harbor Perspectives in Medicine, v.7, n.4, p. 1-12, 2017.
KAMIHARA, J.; HANA, H.Q.; GARBER, J.E. Germline TP53 Mutations and the Changing Landscape of Li-Fraumeni Syndrome. Human Mutation, v. 35, n. 6, p. 654-662, 2014.
KASTAN, M. B. et al. Participation of p53 protein in the cellular response to DNA damage. Cancer research, v. 51, n. 23 Part 1, p. 6304-6311, 1991.
KAVALEC, F. L. Proliferaçăo e Apoptose de Fibroblastos com Mutaçăo no Gene TP53 de Paciente com Síndrome de Li-Fraumeni. 2006.
KERN, S. E. et al. Identification of p53 as a sequence-specific DNA-binding protein. Science, v. 252, n. 5013, p. 1708-11, Jun 21 1991.
KUBBUTAT, Michael HG; JONES, Stephen N.; VOUSDEN, Karen H. Regulation of p53 stability by Mdm2. Nature, v. 387, n. 6630, p. 299, 1997.
LALLOO, F.; VARLEY, J.; ELLIS, D. et al. Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. The Lancet, v. 361, p.1101-1102, 2003.
LEVINE, A. J.; OREN, M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer, v. 9, n. 10, p. 749-58, Oct 2009.
LI, F.P.; FRAUMENI, J.F. et al. A Cancer Family Syndrome in Twenty-Four Kindreds. Cancer Research, v.48, p.5358-5362, 1988.
LÓPEZ, A.R; SÁNCHEZ, P.P; SALCEDO, M. Variaciones genéticas del gen supresor de tumors TP53: relevancia y estrategias de analyses. Laboratório de Oncologia Genomica. Hospital de Oncologia. Rincón del Residente. P, 2-11, 2006.
MATHIAS, C. Análise da Mutação R337H TP53 em Pacientes com Carcinomas Mamários Esporádicos. P, 9-34, 2014.
OLAZAR, M. R. R. Uma metodologia para a descoberta de marcadores genéticos em estudos de associação. 2013. 149f. Tese Doutorado - Universidade Federal do Rio de Janeiro.
OLIVIER, M.; HOLLSTEIN, M.; HAINAUT, P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspectives in Medicine, v. 2, n. 1, p.1-17, 2010.
PICKSLEY, S. M.; LANE, D. P. The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control by p53? Bioessays, v. 15, n. 10, p. 689-90, Oct 1993.
PINTO, E.M.; BILLERBECK, A.E.C.; VILLARES, M.C.B.F. et al. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arquivos Brasileiros de Endocrinologia & Metabologia, v. 48, n. 5, p. 647-650, 2004.
PIOVEZAN, G.C. Prevalência do Alelo TP53 R337H no Estado do Paraná. Tese de Mestrado. Universidade Federal do Paraná, 2006.
RIBEIRO, R.C.; SANDRINI, F.; FIGUEIREDO, B. et al. An Inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proceedings of the National Academy of Sciences of the United States of America, v. 98, n. 16, p. 9330-9335, 2001.
RIDLEY, M. Evolução. Eitora Artmed- 3°ed. P. 170-172. Porto Alegre, 2006.
ROSSNER, P. J.; GAMMON, M.D.; ZHANG, Y-J. et al. Mutation in p53, p53 protein overexpression and breast cancer survival. Journal of Cellular and Molecular Medicine, v. 13, n. 9B, p. 3847-3857, 2009.
ROSSI, C. et al. Pediatric cancer and Li-Fraumeni/Li-Fraumeni-like syndromes: a review for the pediatrician. Revista da Associação Médica Brasileira (1954), v. 61, no. 3 (Maio/Jun. 2015), p. 282-289, 2015.
SHERR C. J. Cancer cell cycles. Science, 274: 1672-1677, 1996.
WANG, Shulin; EL-DEIRY, Wafik S. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene, v. 22, n. 53, p. 8628, 2003.
VARLEY, J.M.; EVANS, D.G.; BIRCH, J.M. Li-Fraumeni syndrome - a molecular and clinical review. British Journal of Cancer, v. 76, n. 1, 1997a.
VARLEY, J.M.; McGOWN, G.; THORNCROFT, M. et al. Germ-line Mutations of TP53 in Li-Fraumeni Families: An Extended Study of 39 Families. Cancer Research, v. 57, n. 15, p 3245-3252, 1997b.
YAN, W.; CHEN, X. Characterization of Functional Domains Necessary for Mutant p53 Gain of Function. The Journal of Biological Chemistry, v. 285, n. 19, p. 14229-14238, 2010.